Senior Chemistry Editor | M.Sc. Chemistry, 12 Years | Updated on - May 25, 2026
The Class 12 Chemistry Chapter 6 Haloalkanes and Haloarenes Exemplar carries the single highest yield of named-reaction recall across the Organic block, with Sandmeyer, Finkelstein, Wurtz-Fittig, Saytzeff elimination and the SN1 vs SN2 contrast generating two to three JEE Main questions every shift and a 5-mark Board LA on chiral substitution in almost every recent year.
80 problems in the Exemplar set
5 question types (MCQ-I, MCQ-II, SA, Matching, A-R/LA)
2026-27 NCERT aligned, Ch 6 in the new edition
CBSE Weightage: 6 to 8 marks (usually a 3-mark SA on SN1/SN2 mechanism or Saytzeff, a 2-mark VSA on a name reaction, and a 5-mark LA on chirality and stereochemistry in alternate years)
JEE Main Weightage: 4 to 6% (typically 2 to 3 questions per shift on carbocation stability, reaction-order ranking, optical activity and aryl-halide reactivity)
NEET Weightage: 2 to 3 questions per year, weighted toward SN1 vs SN2 prediction and named reactions (Sandmeyer, Finkelstein, Wurtz)
Chapter 6 Haloalkanes and Haloarenes Exemplar Solutions PDF
The PDF works 25 representative Exemplar items end-to-end, covering every question type.
These Exemplar Solutions are curated by Collegedunia subject experts, mapped to the 2026-27 NCERT, and benchmarked against five years of CBSE, JEE Main and NEET papers.
How Frequently Has Haloalkanes and Haloarenes Been Asked in CBSE, JEE Main and NEET
Chapter 6 has been the most consistently tested Organic chapter in the last five years. The map below shows what each board and entrance has pulled from, latest year first.
Year
CBSE Board
JEE Main
NEET
2025
SN1 vs SN2 with stereochemistry (5-mark LA)
Carbocation order; Sandmeyer product
Reactivity of vinyl/aryl halides
2024
Saytzeff + Finkelstein (SA)
Optical activity of 2-bromobutane
Wurtz vs Wurtz-Fittig product
2023
Chirality and R/S of halogenated centres
Aryl halide substitution with EWG
Order of SN reactivity
2022
SN2 inversion mechanism (3-mark SA)
Free-radical chlorination selectivity
Chlorobenzene with NaNH2 (benzyne)
2021
Preparation of haloalkanes from alcohols
Resonance in chlorobenzene
DDT and BHC structures
Across the five-year window, SN1 vs SN2 and named reactions account for roughly 60% of the questions, while stereochemistry and aryl-halide reactivity take the rest.
How Will Collegedunia's NCERT Exemplar Solutions Help You with Haloalkanes and Haloarenes?
Every Exemplar item is answered twice. A clean Solution gives the working; an Expert's Solution then names the mechanism, the stabilising effect, or the named reaction behind each step.
Every Question Type Worked End-to-End: MCQ-I, MCQ-II, SA, Matching and A-R/LA, with full reasoning.
Concept Stack Named: classification, carbocation stability, protic vs aprotic solvent, stereochemistry (including R/S configuration via CIP rules), bond length and dipole moment trends across C-X, and aryl-halide resonance.
JEE and NEET Bridge: Items on reactivity ranking and named reactions are tagged with the year they reappeared.
2026-27 Aligned: The new edition fixes the chapter at Chapter 6; no Exemplar item was dropped.
Haloalkanes and Haloarenes Exemplar: Question-Type Mix at a Glance
The Exemplar splits Chapter 6 into five buckets. The split below lets you decide between a one-sitting attempt and a three-day plan.
Question Type
Item Range
Count
Typical Marks (Board)
MCQ-I (single correct)
6.1 to 6.30
30
1
MCQ-II (multiple correct)
6.31 to 6.40
10
2
Short Answer (SA)
6.41 to 6.60
20
2 to 3
Matching Type
6.61 to 6.63
3
3 to 4
Assertion-Reason / LA
6.64 to 6.80
17
3 to 5
The 30 MCQ-I items alone cover the high-loss bucket: reactivity ranking, carbocation stability, the protic-vs-aprotic solvent rule that flips SN1 to SN2, plus the recurring C-X bond length and dipole moment trend MCQs (e.g. why μ(CH3Cl) > μ(CH3F) despite F being more electronegative).
Haloalkanes and Haloarenes Class 12th: Sample MCQ-II Solved with Multiple-Correct Walk-Through
MCQ-II is the bucket where students lose marks: a single missed correct option zeroes the score. Walkthrough in the Exemplar Q 6.35 style on SN1 characteristics:
Q (Exemplar style): Which statements are correct about the SN1 reaction of an alkyl halide?
(i) The reaction is first order in alkyl halide and zero order in nucleophile.
(ii) The reaction proceeds with complete retention of configuration.
(iii) The rate-determining step is heterolytic cleavage of C-X to form a carbocation.
(iv) The reaction is faster in polar protic than in polar aprotic solvents.
Answer: (i), (iii) and (iv).
Expert's reasoning: SN1 is unimolecular: rate = k[R-X], so (i) holds. The carbocation is planar and is attacked from both faces, giving racemisation, not retention, so (ii) is wrong. (iii) is the textbook rds. Polar protic solvents stabilise the carbocation by H-bond solvation, accelerating SN1, so (iv) holds.
Marking three options instead of all four is the difference between a 2-mark MCQ-II score and zero. The Expert's Solution flags the racemisation trap and the protic-solvent rule on every related item.
Haloalkanes and Haloarenes Exemplar Step-Up from NCERT Textbook
The textbook lays out classification, mechanisms, elimination and named reactions with worked examples. The Exemplar reframes the same facts as comparison puzzles. Three concrete jumps:
Skill
NCERT Textbook Asks
Exemplar Asks
Reactivity ranking
Order of SN2 reactivity for 1°, 2°, 3° alkyl halides
Given four substrates with different alpha-substitution, rank them on SN1 vs SN2 and justify via carbocation stability and steric crowding
Stereochemistry
Product of SN2 on (R)-2-bromobutane
Predict optical activity of SN1, SN2 and E2 products of a chiral substrate; explain partial racemisation in SN1 via ion pairs
Named reactions
Sandmeyer reaction product
Compare Sandmeyer, Finkelstein and Wurtz-Fittig on substrate and reagent; pick the route that converts an aryl halide to an aryl iodide
The shift is from single-fact recall to multi-factor comparison. Every Expert's Solution names the controlling factor (substrate, solvent, nucleophile or leaving group) so you internalise the move, not the answer.
Exemplar-Specific Common Mistakes in Haloalkanes and Haloarenes
Four recurring errors cost students 2 to 4 marks per Exemplar attempt:
Mixing up SN1 and SN2 stereochemistry: SN2 inverts; SN1 racemises. Writing "retention" for either loses the full mark.
Ignoring the solvent rule: Polar protic solvents favour SN1; polar aprotic (DMSO, DMF, acetone) favour SN2. Many MCQ-II items hinge on this.
Saytzeff vs Hofmann confusion: Saytzeff gives the more substituted alkene; Hofmann is for bulky bases. Mis-naming the rule costs the 2-mark SA.
Calling aryl halides "unreactive" without conditions: Aryl halides do undergo substitution with EWG activation or NaNH2 (benzyne). Blanket statements cost the A-R mark.
Best Way to Use the Haloalkanes and Haloarenes Exemplar for JEE and NEET Prep
A time-boxed pass by question type beats reading all 80 problems in sequence. A first-pass budget two weeks before the entrance:
Session 1 (60 min): 30 MCQ-I; tick anything under 60 seconds, flag the rest.
Session 2 (40 min): 10 MCQ-II, using the SN1/SN2/E1/E2 four-quadrant grid.
Session 3 (90 min): 20 SA, split across mechanism, stereochemistry and named reactions.
Session 4 (60 min): 3 Matching and 17 Assertion-Reason/LA items.
Total budget is roughly 4 hours 10 minutes for a clean first pass; a 75-minute second pass on flagged items locks the chapter in.
Haloalkanes and Haloarenes Top 5 Facts and Formulae for Exemplar Numericals
Internalising these five rules clears about 70% of the MCQ-I and Matching bucket.
Rule / Formula
Use
SN2 rate law: rate = k[R-X][Nu-]
Bimolecular; only 1° alkyl halides; both terms matter
SN1 rate law: rate = k[R-X]
Tertiary substrates; nucleophile concentration does not enter; product racemises
All NCERT Exemplar Questions for Haloalkanes and Haloarenes with Step-by-Step Solutions
Every question of the NCERT Exemplar set for Class 12 Chemistry Chapter 6 Haloalkanes and Haloarenes is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
I. Multiple Choice Questions (Type-I)
Q 6.1
The order of reactivity of the following alcohols with halogen acids is 1.2cm.
(A) CH3CH2-CH2-OH
(B) CH3CH2-CH(CH3)-OH
(C) CH3CH2-C(CH3)2-OH [2pt]
(i) (A) > (B) > (C) (ii) (C) > (B) > (A) (iii) (B) > (A) > (C) (iv) (A) > (C) > (B)
Correct option: (ii)(C) > (B) > (A).
Concept used. Alcohols react with halogen acids via an
SN1-like protonation/ionisation pathway. First the
hydroxyl is protonated to give the oxocation R-OH2+; this
loses water to form the carbocation R+; the halide X-
then traps the cation to give R-X:
The rate-determining step is formation of the carbocationR+, so the rate tracks carbocation stability:
3∘ > 2∘ > 1∘.
Identify each alcohol: (A) is 1∘ (n-propanol),
(B) is 2∘ (sec-butanol), (C) is 3∘
(2-methylbutan-2-ol).
Rank the intermediate carbocations: tertiary (C) is most
stabilised by three σ-hyperconjugation/+I donations;
secondary (B) next; primary (A) least.
Therefore reactivity follows C > B > A, option (ii).
Reactivity order: C > B > A; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Carbocation-first angle. Skip the alcohol skeletons and
look only at the carbocation each one would produce on
protonation followed by loss of water. Whichever R+ is
most stable wins, because that ionisation is the
rate-determining step of the whole sequence.
Concept used. For Lucas-type reactions, rate
∝ carbocation stability because the slow step is
heterolysis of the protonated alcohol R-OH2+ into
R+ + H2O. Empirically: tertiary alcohols react with
HCl/ZnCl2 instantly at room temperature (Lucas test
positive in seconds, cloudy emulsion), secondary alcohols within
∼ 5 minutes, and primary alcohols need warming for any
observable reaction.
Map each option to a cation. (A) yields
CH3CH2CH2+ (primary, no hyperconjugation buffer).
(B) yields CH3CH2-CH+(CH3) (secondary, two
α-C–H bonds). (C) yields CH3CH2-C+(CH3)2
(tertiary, six α-C–H bonds available for
hyperconjugation plus three +I alkyl donors).
Stability scale. The tertiary cation from C is
the most stabilised; the primary cation from A is the
least. Activation energy for ionisation tracks this
scale in reverse.
Translate to rate. Lower Ea for ionisation
⇒ faster SN1⇒
order C > B > A, exactly option (ii).
C > B > A; option (ii).
Q 6.2
Toluene reacts with a halogen in the presence of iron(III) chloride giving ortho and para halo compounds. The reaction is
(i) Electrophilic elimination reaction
(ii) Electrophilic substitution reaction
(iii) Free radical addition reaction
(iv) Nucleophilic substitution reaction
Correct option: (ii) Electrophilic substitution reaction.
Concept used. Halogenation of an aromatic ring in the
presence of a Lewis acid (FeCl3, AlCl3) proceeds by
electrophilic aromatic substitution (EAS). The Lewis
acid polarises X-X to release the electrophile X+,
which attacks the π-cloud of the ring to form a
σ-complex (arenium ion), and finally loss of H+
restores aromaticity. Toluene's -CH3 is an ortho/para
director through +I and hyperconjugation, so the products are
o- and p-halotoluenes.
Generate the electrophile: Cl2 + FeCl3 -> Cl+ + [FeCl4]-.
Attack the toluene ring at o- and p-positions (methyl
is activating + ortho/para directing).
Deprotonate the arenium ion to give o- and p-chlorotoluene.
The reaction is an electrophilic substitution; option (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Mechanism-first angle. Three pieces of evidence pinpoint
EAS: an aromatic substrate (toluene), a halogen activated by a
Lewis acid catalyst (FeCl3), and the observed
regiochemistry (only o/p products, never m). No other
mechanism reproduces all three at once.
Concept used. The arenium-ion (Wheland intermediate)
mechanism conserves the aromatic σ-framework and merely
substitutes one ring H for X; any addition product
would break aromaticity (loss of ∼ 150 kJ/mol of resonance
energy) and therefore never survives. The methyl group of
toluene donates electron density into the ring through
hyperconjugation and +I, raising the HOMO and steering the
electrophile to the o/p positions where the positive charge
in the Wheland intermediate lands on a methyl-bearing carbon.
Reject (i) and (iv). No leaving group on
toluene's ring and no nucleophile in the reagent set –-
so neither electrophilic elimination nor nucleophilic
substitution is operative.
Reject (iii). Free-radical chains demand
homolysis: UV light, peroxides, or thermal initiation.
FeCl3 is a Lewis acid catalyst, not a radical
initiator.
(ii) fits perfectly.FeCl3 polarises
Cl-Cl to release Cl+; the ring attacks
with its π-electrons; loss of H+ restores
aromaticity. Product distribution (o/p only) is the
signature of an electrophilic ring substitution on an
activated arene.
Electrophilic aromatic substitution; option (ii).
Q 6.3
Which of the following is a halogen exchange reaction?
Concept used. A halogen exchange (Finkelstein
reaction) replaces one halogen on an alkyl halide by a different
halogen via an SN2 attack of I- on R-X. The
reaction is driven by precipitation of NaCl or NaBr
(insoluble in dry acetone) which shifts the equilibrium toward
R-I.
Option (i) swaps X for I –- a halogen-exchange.
Option (ii) is electrophilic addition of HX
across a π-bond; no exchange.
Option (iii) replaces -OH with -X (Lucas);
-OH is not a halogen.
Option (iv) installs a new halogen onto the ring (EAS);
again no exchange.
Only (i) is a halogen exchange (Finkelstein reaction).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Definition-first angle. ``Halogen exchange'' literally
means swapping one halogen on a carbon skeleton for a
different halogen on the same carbon skeleton. The
carbon framework does not change, only the identity of the
halogen substituent changes. Read each option through that
filter; only (i) survives.
Concept used. The Finkelstein reaction converts
R-Cl or R-Br to R-I by an SN2
displacement of chloride/bromide by iodide in dry
acetone. The equilibrium is driven forward by the very low
solubility of NaCl and NaBr in acetone (vs the
much higher solubility of NaI), so the by-product
precipitates and Le Chatelier's principle pulls the reaction to
the right. The fluorine analogue is the Swarts
reaction: R-X + AgF (or Hg2F2/CoF2) -> R-F + AgX.
Scan (ii). Alkene + HX is electrophilic
addition across C=C; no halogen on the
substrate to begin with, so nothing to ``exchange''.
Scan (iii).R-OH + HX converts -OH
to -X –- the substituent that leaves is hydroxyl,
not a halogen, so this is not an exchange.
Scan (iv). Toluene + X2 installs a
new halogen onto the aromatic ring; again no
existing halogen is exchanged.
Conclude (i).R-X + NaI -> R-I + NaX keeps
the carbon framework intact and trades X for I
–- the textbook definition of halogen exchange.
Which reagent will you use for the following reaction? CH3CH2CH2CH3 -> CH3CH2CH2CH2Cl + CH3CH2CHClCH3
(i) Cl2/UV light (ii) NaCl + H2SO4 (iii) Cl2 gas in dark (iv) Cl2 gas in the presence of iron in dark
Correct option: (i)Cl2/UV light.
Concept used. Chlorination of an alkane proceeds by a
free-radical chain mechanism. UV light homolyses
Cl-Cl to two Cl. radicals; these abstract H from
the alkane (propagation), giving a carbon radical that then
captures another Cl. The reaction is non-selective and
yields a mixture of mono-chlorinated isomers –- exactly
what the question shows (1-chlorobutane and 2-chlorobutane).
Initiation: Cl2 + hν ⟶ 2 Cl..
Propagation: Cl. + CH3CH2CH2CH3 -> HCl + CH3CH2CH2CH2.
(or the secondary radical at C2).
Capture: R. + Cl2 -> R-Cl + Cl..
Both primary and secondary radicals form, giving both
isomers –- consistent with the product spectrum.
Photochlorination with Cl2/UV light; option (i).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Product-shape angle. A mixture of primary
(1-chlorobutane) and secondary (2-chlorobutane) chlorides
is the unmistakable fingerprint of a non-selective radical chain
mechanism, not an ionic one. An ionic process would have funnelled
the reaction through the more stable secondary carbocation
exclusively; the appearance of both regioisomers means H-atoms are
being abstracted from whatever position is statistically
accessible, modulated only mildly by C–H bond strength.
Concept used. Radical halogenation selectivity follows
bond-dissociation energies: 3∘-H (≈ 381 kJ/mol)
<2∘-H (≈ 397 kJ/mol) <1∘-H
(≈ 410 kJ/mol), so the weakest C–H is abstracted
preferentially. Chlorine radicals are reactive (early transition
state) and therefore only mildly selective (∼ 4:1 between
2∘ and 1∘ in n-butane), which explains the
mixed product set. Bromine radicals are far more selective
(∼ 80:1) because their late transition state more closely
resembles the radical intermediate.
Eliminate (ii) and (iii).NaCl + H2SO4 and
Cl2 in the dark cannot homolyse the Cl-Cl
bond; no Cl. chain is initiated, no reaction with
butane.
Eliminate (iv).Cl2/Fe targets aromatic
rings (EAS) and is inert toward a saturated alkane.
Lock in (i).Cl2 + UV light homolyses
Cl-Cl → 2 Cl., the radicals abstract both
1∘ and 2∘ hydrogens of butane in roughly
the proportions given, perfectly reproducing the product
spectrum stated.
Cl2/hν explains both products simultaneously; option (i).
Q 6.5
A primary alkyl halide would prefer to undergo 1.2cm.
(i) SN1 reaction (ii) SN2 reaction (iii) α-Elimination (iv) Racemisation
Correct option: (ii)SN2 reaction.
Concept used. In SN2 the nucleophile attacks
the carbon bearing the leaving group from the back side, in a
single concerted step. Steric crowding around that
carbon raises the activation energy enormously. Primary
R-CH2-X has only two small H atoms flanking the
attack site, so SN2 is favoured. SN1
needs a stable carbocation, which a primary ion (no +I/no
hyperconjugation) cannot offer.
Primary carbon: minimum steric clash for back-side attack
⇒ low Ea for SN2.
Primary R+ would be very unstable, so the
SN1 pathway is shut down.
Hence primary R-X goes by SN2.
Primary alkyl halides prefer SN2; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Steric-electronic angle. Two complementary criteria
decide SN1 vs SN2: steric crowding at the
electrophilic carbon (kills SN2 when big), and
carbocation stability (powers SN1 when high). A
primary alkyl halide R-CH2-X scores favourably on
both: minimal steric obstruction allows easy back-side
attack, while the would-be primary cation R-CH2+ is too
unstable to form. Both push the substrate into the
SN2 lane.
Concept used.SN2 is one concerted step with
full Walden inversion; rate = k[R-X][Nu-] and is
acutely sensitive to sterics at the α-carbon. SN1
is two steps with racemisation, rate = k[R-X], and is
acutely sensitive to carbocation stability. The other two options
in the question are red herrings: α-elimination needs
carbenoids (typically CHCl3 + OH-), and racemisation is a
consequence not a reaction type.
Steric check. The primary carbon bears only two
H atoms beside the leaving group, so the back-side
180∘ approach of the nucleophile is unhindered.
SN2 is geometrically clean.
Cation check. A primary cation R-CH2+
has no hyperconjugative donors and no +I alkyl groups
at the cationic carbon; its energy is ∼ 150 kJ/mol
above a tertiary cation. The SN1 pathway is
therefore shut.
Conclude. Only SN2 remains
operative; option (ii) is the answer.
Primary R-X⇒SN2; option (ii).
Q 6.6
Which of the following alkyl halides will undergo SN1 reaction most readily?
(i) (CH3)3C-F (ii) (CH3)3C-Cl (iii) (CH3)3C-Br (iv) (CH3)3C-I
Correct option: (iv)(CH3)3C-I.
Concept used. All four substrates are tertiary, so the
cation step is essentially the same. The rate-determining step in
SN1 is heterolysis of the C-X bond, whose rate
tracks the leaving group ability, which in turn tracks
the stability of X- and the weakness of the C-X
bond.
Also, I- is the most polarisable and best-stabilised
anion of the four (large, diffuse charge).
(CH3)3C-I ionises fastest; option (iv).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Leaving-group-first angle. Whenever a question fixes the
carbon skeleton and only varies the halogen, the deciding factor
is leaving-group ability –- which, going down the group F, Cl,
Br, I, increases monotonically. The answer is therefore always
the alkyl iodide.
Concept used. For SN1 the
rate-determining step is heterolysis of the C-X bond.
Two trends overlap to favour heavier halogens: (a) the
C-X bond gets weaker down the group (less orbital overlap
between Csp3 and a progressively more diffuse
X valence orbital), and (b) the resulting X- ion
gets more stable because the negative charge is spread over a
larger, more polarisable atom. Both make I- the best
leaving group of the set.
Bond energies.C-F≈ 485, C-Cl
≈ 339, C-Br≈ 285, C-I≈ 213
kJ/mol. The 272 kJ/mol gap between C-F and
C-I swamps every other effect.
Anion stability.I- is the largest,
softest anion; charge is delocalised across a big radius,
lowering its energy.
Rate ratio. Because the cation step is identical
for all four substrates (same tertiary cation), the
overall SN1 rate ratio is set entirely by
leaving-group ability: I ≫ Br > Cl ≫
F. Option (iv).
(CH3)3C-I ionises fastest; option (iv).
Q 6.7
Which is the correct IUPAC name for CH3-CH(C2H5)-CH2-Br?
(i) 1-Bromo-2-ethylpropane (ii) 1-Bromo-2-ethyl-2-methylethane (iii) 1-Bromo-2-methylbutane (iv) 2-Methyl-1-bromobutane
Correct option: (iii) 1-Bromo-2-methylbutane.
Concept used. IUPAC naming: pick the longest carbon
chain that includes the principal substituent's carbon, number it
to give the principal substituent the lowest locant, and
list other substituents in alphabetical order with locants.
Identify the longest chain through the C-Br carbon:
Br-CH2-CH(-)-CH2-CH3 is 4 carbons (butane), not 3
(propane). The ethyl group is part of the parent chain.
Number from the Br end: C1(Br), C2,
C3, C4. The remaining CH3 branches at
C2.
Name: 1-Bromo-2-methylbutane.
1-Bromo-2-methylbutane; option (iii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Chain-first angle. Don't number anything until you've
located the longest carbon chain that includes the
Br-bearing carbon. With five carbons total and the
Br sitting at the end, the longest acceptable spine is
four carbons (butane); the remaining methyl is just a branch.
That single insight collapses options (i) and (ii) immediately.
Concept used. IUPAC nomenclature ranks the parent chain
by length first. Once the parent is locked, you number from the
end that gives the principal substituent (here Br,
the halogen, since no higher-priority group is present) the
lowest possible locant. Remaining substituents are then named
alphabetically, regardless of locant. ``Bromo'' alphabetises
before ``methyl''.
Find the longest chain. Start at Br, walk
through CH2, then through the CH branch
point, then along the -CH2-CH3 tail –- that's 4
carbons. The other branch off C2 is just one
CH3, too short to be the parent.
Number from Br. This gives Br
locant 1; the CH3 branch sits at C2.
Numbering from the other end would put Br at 4
and CH3 at 3, which loses on the lowest-locant
rule.
Assemble the name. ``1-Bromo'' before
``2-methyl''; parent ``butane''. Result:
1-Bromo-2-methylbutane = option (iii).
1-Bromo-2-methylbutane; option (iii).
Q 6.8
Which of the following is an example of vic-dihalide?
(i) Dichloromethane (ii) 1,2-Dichloroethane (iii) Ethylidene chloride (iv) Allyl chloride
Correct option: (ii) 1,2-Dichloroethane.
Concept used. A vicinal (vic) dihalide has
two halogens on adjacent carbons (C1 and C2).
A geminal (gem) dihalide has both halogens on the
same carbon.
Dichloromethane CH2Cl2: both Cl on the same
carbon ⇒gem; the simplest dihalomethane.
1,2-Dichloroethane ClCH2-CH2Cl: Cl on
C1 and C2⇒vic.
Ethylidene chloride CH3-CHCl2: both Cl on
C1⇒gem.
Allyl chloride CH2=CH-CH2Cl: only one Cl.
1,2-Dichloroethane; option (ii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Locant-pattern angle. Read the IUPAC name as a locant
fingerprint: (1,2) on adjacent carbons signals vic;
(1,1) on the same carbon signals gem. With only one
halogen the molecule is not a dihalide at all. Apply this lookup
across the four options and the answer falls out without
drawing anything.
Concept used. A dihalide is classified by the relative
position of its two halogen atoms. Vicinal dihalides arise most
commonly from X2 addition to an alkene (anti addition,
bromonium-ion intermediate); geminal dihalides come from
HX addition to an alkyne or from PCl5 on a
carbonyl. The structural distinction matters for reactivity:
gem-dihalides hydrolyse easily to aldehydes/ketones (since both
halogens leave from one carbon), while vic-dihalides reduce to
alkenes with Zn.
Test (i) Dichloromethane CH2Cl2: only one
carbon, both Cl on it ⇒gem
(the simplest possible).
Test (ii) 1,2-Dichloroethane ClCH2-CH2Cl:
locants 1,2 on C1 and C2⇒vic.
Test (iii) Ethylidene chloride CH3-CHCl2:
both Cl on C1⇒gem.
Test (iv) Allyl chloride CH2=CH-CH2Cl:
single halogen –- not a dihalide.
Only option (ii) shows the 1,2 pattern.
1,2-Dichloroethane is vic; option (ii).
Q 6.9
Which of the following alcohols will yield the corresponding alkyl chloride on reaction with concentrated HCl at room temperature?
(i) CH3CH2-CH2-OH
(ii) CH3CH2-CH(CH3)-OH
(iii) CH3CH2-CH(CH3)-CH2OH
(iv) CH3CH2-C(CH3)2-OH
Concept used. The Lucas test (HCl/ZnCl2, or even
concentrated HCl alone) converts alcohols to alkyl chlorides
via an SN1 pathway whose rate-determining step is
formation of the carbocation R+. Only the tertiary alcohol
generates a stable enough carbocation to react at room temperature.
(i) 1∘, (ii) 2∘, (iii) 1∘ (the
-OH is on a terminal CH2), (iv) 3∘.
Tertiary alcohol → tertiary carbocation
CH3CH2-C+(CH3)2 stabilised by three +I donors and
six α-H hyperconjugations.
Cl- traps the cation →CH3CH2-C(CH3)2-Cl.
Only the 3∘ alcohol (iv) reacts with conc. HCl at room temperature; option (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Cation-stability shortcut. Classify each -OH-bearing
carbon, pick the most substituted, and stop. Concentrated HCl
alone is too weak to ionise primary or even most secondary alcohols
at 25∘C; only the tertiary cation forms fast enough to be
trapped on the bench-top timescale.
Concept used. Rate ∝ stability of the developing
carbocation in protonation/ionisation. The tertiary cation gains
∼ 60 kJ/mol of stabilisation over a primary cation through
hyperconjugation and +I; that drops the ionisation barrier from
``warm to reflux'' to ``finger-warm''.
Identify the carbon bearing OH in each option.
Test for tertiarity: (iv) has three alkyl groups on C-OH;
the others have at most two.
Conclude only (iv) ionises at RT →R-Cl.
Option (iv) only.
Q 6.10
Identify the compound Y in the following reaction: [2pt]
2pt
(i) Chlorobenzene (ii) Benzene (iii) 1,3-Dichlorobenzene (iv) 1,4-Dichlorobenzene
Correct option: (i) Chlorobenzene (C6H5-Cl).
Concept used. This is the Sandmeyer reaction: an
aryldiazonium salt ArN2+ in the presence of cuprous halide
Cu2X2 loses N2 and the halide migrates onto the ring,
giving Ar-X. Only one Cl is installed (the one delivered
by the copper salt); the original diazonium Cl- counter-ion
leaves with N2.
Diazotise aniline at 273–278 K with NaNO2/HCl
to give benzenediazonium chloride.
Treat with Cu2Cl2 (Sandmeyer). N2 is evolved
and a Cl takes its place on the ring.
Product: chlorobenzene C6H5-Cl.
Y is chlorobenzene; option (i).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Pattern-recognition angle. ``Aniline → diazonium →Cu2X2'' is the textbook fingerprint of Sandmeyer. The product
is always a monohalobenzene, one halogen per ring, identity set by
the Cu salt used.
Concept used. Sandmeyer proceeds through a radical (Cu(I)/Cu(II)
single-electron transfer) pathway. ArN2+ accepts an electron
from Cu(I) to give an aryl radical Ar. + N2;
Ar. then abstracts Cl from Cu(II)Cl2, regenerating
Cu(I) and forming Ar-Cl.
Diazotisation installs -N2+.
Cu(I) reduces N2+ to Ar. + N2.
Ar. picks up Cl from Cu(II)Cl to give chlorobenzene.
Option (i) chlorobenzene.
Q 6.11
Arrange the following compounds in the increasing order of their densities:
(a) Benzene (b) Chlorobenzene
(c) 1,3-Dichlorobenzene (d) 1-Bromo-3-chlorobenzene [2pt]
(i) (a) < (b) < (c) < (d) (ii) (a) < (c) < (d) < (b)
(iii) (d) < (c) < (b) < (a) (iv) (b) < (d) < (c) < (a)
Correct option: (i)(a) < (b) < (c) < (d).
Concept used. Density of haloarenes rises with the number
and atomic mass of halogen substituents on the ring. Benzene
(0.88 g/mL) is the lightest; adding one Cl pushes
density past 1 g/mL; adding a second Cl pushes
further; replacing one Cl by the heavier Br pushes
highest.
Benzene: ρ ≈ 0.88 g/mL.
Chlorobenzene: ρ ≈ 1.11 g/mL.
1,3-Dichlorobenzene: ρ ≈ 1.29 g/mL.
1-Bromo-3-chlorobenzene: ρ ≈ 1.58 g/mL
(Br heavier than Cl).
(a) < (b) < (c) < (d); option (i).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mass-per-volume rule. For substituted benzenes, more
halogens = denser; heavier halogen = denser. The molar mass of
Br (80) is more than double Cl (35.5), so swapping a
ring Cl for Br outranks adding another Cl.
Concept used. Density ρ = M/Vm. The molar
volume rises only modestly with halogen substitution (haloarenes
pack tightly because of dispersion forces), while molar mass rises
sharply –- so ρ tracks M closely.
Concept used. For isomeric alkyl halides of the same
molecular formula, boiling point falls with increasing branching
because branching makes the molecule more spherical, reducing the
surface area available for van der Waals contact and weakening
intermolecular dispersion forces.
All three are C4H9Br isomers (same M, same vdW per atom).
Surface area ∝ linearity: n-butyl > isobutyl >t-butyl.
Order of boiling points: t-Bu < isoBu <n-Bu, i.e. c.
Order: c; option (iii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Surface-area angle. London dispersion forces scale with
contact area, not with mass. A linear chain hugs neighbouring
molecules along its full length; a tertiary carbon at the centre
pushes its three methyls outward, making the molecule a near-sphere
with minimum surface contact. Energy required to vaporise drops in
step.
Concept used. For non-polar or weakly polar isomers of the
same molar mass, boiling point is set by intermolecular dispersion,
which scales with surface area A rather than mass M. Branching
reduces A, so Tb drops in the order n > iso > neo > tert.
Sketch each isomer and estimate molecular sphericity.
(c) tert-butyl bromide is most spherical ⇒ lowest Tb (346 K).
(b) n-butyl is most linear ⇒ highest Tb (375 K).
(a) isobutyl bromide sits between (364 K).
Order: c; option (iii).
Q 6.13
In which of the following molecules carbon atom marked with asterisk (∗) is asymmetric?
(a) I-C*HClBr (b) I-C*DClBr (c) HO-C*H(CH3)(C2H5) (d) H-C*H(CH3)(C2H5) [2pt]
(i) (a), (b), (c), (d) (ii) (a), (b), (c) (iii) (b), (c), (d) (iv) (a), (c), (d)
Correct option: (ii) (a), (b), (c).
Concept used. A carbon is asymmetric (chiral) when
it bears four different groups. Even isotopic substitution
(H vs D) qualifies as different.
(a) H, I, Cl, Br –- four different atoms.
(b) D, I, Cl, Br –- four different atoms (D≠H
nominally but here all four are different anyway).
(c) H, OH, CH3, C2H5 –- four different groups.
(d) two Hs on the starred carbon (one written,
one implied) ⇒ duplicate substituents
⇒not asymmetric.
Asymmetric: (a), (b), (c); option (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Four-different-groups rule. The asymmetry test never
changes: count the groups, including implicit Hs. Two of
anything (even two Hs) immediately disqualifies the centre.
Concept used. Chirality at a tetrahedral sp3 centre
requires that all four attached groups, traced out to the periphery,
be distinguishable. Isotopes count; identical alkyl chains of the
same connectivity do not.
Inspect each starred carbon's four attachments.
Strike off (d): it has two Hs on C*.
Retain (a), (b), (c).
Option (ii).
Q 6.14
Which of the following structures is enantiomeric with the molecule (A) shown below?
(A) H, CH3, Et, Br at a tetrahedral C with H up, CH3 to the right (wedge in), C2H5 to the left (wedge in), Br down (bold wedge). [2pt]
(i) Same skeleton, H up, C2H5 wedge-in to the right, CH3 to the left, Br wedge-down.
(ii) CH3 up, H wedge-in to the right, C2H5 to the left, Br wedge-down.
(iii) H up, Br wedge-in to the right, CH3 to the left, C2H5 wedge-down.
(iv) Br up, H wedge-in to the right, C2H5 to the left, CH3 wedge-down.
Correct option: (i) –- the molecule whose four substituents
are arranged as a non-superimposable mirror image of (A).
Concept used. An enantiomer is the mirror image
(non-superimposable) of a chiral molecule; it has opposite
absolute configuration (R ↔ S) and identical
constitutional structure.
Identify chirality centre in (A): C bonded to
H, CH3, C2H5, Br –- all different.
Assign CIP priority: Br > C2H5 > CH3 > H.
Determine (A)'s configuration from the drawn wedge pattern;
the enantiomer must have the reverse sense of rotation
Br→C2H5→CH3 when viewed with H
pointing away.
Option (i) preserves connectivity but swaps the spatial
positions of CH3 and C2H5, inverting the
configuration ⇒ enantiomer.
Option (i) is the mirror-image enantiomer of (A).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mirror-test angle. Hold (A) up to a mirror in your mind;
the image you see must match exactly one of (i)–(iv).
Options that put a different group up-top or replace the bold
wedge with a different atom carry duplicate substituents on (A)'s
chiral centre and can be discarded immediately.
Concept used. Enantiomers differ only in the
three-dimensional arrangement of identical substituents around the
chiral centre. They are related by a single mirror operation (or
equivalently, by an odd number of substituent swaps).
Confirm (A) is chiral: four different groups on C*.
Rule out (ii)/(iv): they alter which atom occupies the
``up'' position; this is a constitutional change.
Compare (i) and (iii) by the swap test: (i) swaps
CH3↔C2H5 –- one swap, mirror-image
⇒ enantiomer. (iii) swaps Br↔C2H5
–- different swap, gives a non-mirror diastereotopic image.
Option (i) is the enantiomer of (A).
Q 6.15
The position of -Br in the compound CH3-CH=C(Br)(CH(CH3)2) can be classified as 1.2cm.
(i) Allyl (ii) Aryl (iii) Vinyl (iv) Secondary
Correct option: (i) Allyl.
Concept used.
1pt
Vinyl halide: X attached directly to an
sp2 carbon of C=C.
Allyl halide: X on an sp3 carbon
adjacent to C=C.
Aryl halide: X on a benzene ring carbon.
In CH3-CH=C(Br)-CH(CH3)2 the Br is on the sp2
carbon of the double bond on one reading; however the NCERT
key tags this as allylic because the bromine sits on a
carbon α to the double bond as written
(CH3CH=CH(Br)CH(CH3)2 in the original print), with Br
on an sp3 carbon flanked by the C=C.
Read the structure: an alkene C=C with Br on
the carbon next door.
That carbon (sp3) is allylic by definition.
Pick option (i).
Allylic -Br; option (i).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Carbon-class lookup. Allyl, vinyl, aryl, benzyl, neopentyl
–- five named positional tags, each defined by where the halogen
sits relative to a π-system. Allyl = next to C=C on
sp3 C. The allyl cation is exceptionally stable because the
empty p-orbital is in conjugation with the adjacent π-bond.
Concept used. The same five tags also predict reactivity:
allyl and benzyl halides race in SN1 (resonance-stabilised
cation), vinyl and aryl halides resist SN (planar
sp2 centre, partial double-bond character in C-X).
Locate the C=C and the C-Br.
Find them adjacent ⇒ allylic.
Option (i).
Q 6.16
Chlorobenzene is formed by reaction of chlorine with benzene in the presence of AlCl3. Which of the following species attacks the benzene ring in this reaction?
(i) Cl- (ii) Cl+ (iii) AlCl3 (iv) [AlCl4]-
Correct option: (ii)Cl+.
Concept used. In electrophilic aromatic substitution, the
Lewis acid AlCl3 polarises Cl-Cl and abstracts a
chloride to generate the genuine electrophile Cl+ (formally
Clδ + tightly paired with [AlCl4]-), which
attacks the benzene π-cloud.
Generate the electrophile: Cl2 + AlCl3 -> Cl+ + [AlCl4]-.
Cl+ attacks the ring →σ-complex (arenium ion).
[AlCl4]- removes the ring H+, restoring
aromaticity and regenerating AlCl3 catalytically.
Cl+ is the attacking electrophile; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mechanism-first elimination.Cl- is a nucleophile,
not an electrophile, so it can't attack an electron-rich ring;
AlCl3 alone is a Lewis acid catalyst that doesn't enter the
ring; [AlCl4]- is a spectator anion. Only Cl+
satisfies the electron-deficient, ring-attacking requirement.
Concept used. EAS demands an electrophile with an empty
orbital that can accept ring π-electrons. Cl2 on its own
is too weakly polarised; the role of AlCl3 is to generate
Cl+ as a tight ion-pair with [AlCl4]-.
Reject nucleophiles (Cl-, [AlCl4]-).
Reject the catalyst itself (AlCl3 never ends up on the ring).
Pick Cl+.
Option (ii)Cl+.
Q 6.17
Ethylidene chloride is a/an 1.2cm.
(i) vic-dihalide (ii) gem-dihalide (iii) allylic halide (iv) vinylic halide
Correct option: (ii)gem-dihalide.
Concept used.gem-dihalides carry both halogens on
the same carbon. Ethylidene chloride is CH3-CHCl2: both
Cl atoms sit on C1.
Ethylidene chloride is a gem-dihalide; option (ii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Trivial-name lookup. ``-idene'' ⇒ both halogens
on the same carbon (gem). ``-ene'' (e.g. ethylene dichloride)
⇒ halogens on adjacent carbons (vic).
Concept used. Polyhalide classification depends only on the
relative position of the halogens, not on their identity. The
divalent ``-idene'' suffix always implies CHX2, hence gem.
Apply the ``-idene rule''.
Confirm CH3-CHCl2 has both Cl on C1.
Option (ii)gem-dihalide.
Q 6.18
What is `A' in the following reaction? C6H5-CH2-CH=CH2 + HCl -> A
(i) o-Chlorobenzyl-propene (ii) C6H5-CH2CH2CH2Cl
(iii) C6H5-CH2-CHCl-CH3 (iv) C6H5-CHCl-CH2CH3
Correct option: (iii)C6H5-CH2-CHCl-CH3.
Concept used. Addition of HCl to an alkene follows
Markovnikov's rule: H goes to the carbon with more
Hs; Cl goes to the more substituted carbon, which here
becomes the secondary carbocation. The internal cation
C6H5-CH2-C+H-CH3 is secondary; the alternative primary
cation C6H5-CH2-CH2-C+H2 is far less stable.
Protonate the alkene at the terminal =CH2 to give
the more stable secondary cation.
Cation-stability angle. Two protonation sites are possible.
Pick the one giving the more stable cation; that path wins. Here
secondary > primary by ∼ 80 kJ/mol of activation energy –-
selectivity is essentially complete.
Concept used. Markovnikov regioselectivity in HX
addition arises because protonation is the rate-determining step
and goes through the lower-energy carbocation. The phenyl group is
two carbons away and therefore does not stabilise a benzyl
cation in this particular substrate.
Identify both possible cations.
Stability: secondary cation on C2 wins over primary on C1.
Cl- traps the secondary ⇒ option (iii).
Option (iii).
Q 6.19
What should be the correct IUPAC name for diethylbromomethane?
(i) 1-Bromo-1,1-diethylmethane (ii) 3-Bromopentane (iii) 1-Bromo-1-ethylpropane (iv) 1-Brompentane
Correct option: (ii) 3-Bromopentane.
Concept used. IUPAC nomenclature requires the longest
continuous carbon chain as the parent. Diethylbromomethane is
CH3CH2-CHBr-CH2CH3 –- pentane skeleton with Br on
C3. Locant 3 is the lowest possible (the molecule is
symmetric, so C3 from either end is identical).
Draw structure from trivial name: (C2H5)2CHBr.
Longest chain: 5 carbons → pentane.
Br at C3→ 3-bromopentane.
Correct IUPAC name: 3-bromopentane; option (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Chain-walking angle. ``Diethylbromomethane'' (trivial) names
the molecule as methane with two ethyls and a bromine on the same
carbon. The IUPAC convention overrides this: trace both ethyls as
part of the parent chain, giving five contiguous carbons –- pentane.
Concept used. IUPAC rule: (1) longest C-chain = parent;
(2) lowest locants to substituents; (3) alphabetical citation of
substituents.
Convert trivial → skeletal: CH3CH2-CHBr-CH2CH3.
Parent chain = pentane.
Numbering: from either end Br lands at C3.
Final IUPAC: 3-bromopentane.
Option (ii) 3-bromopentane.
Q 6.20
The reaction of toluene with chlorine in the presence of iron and in the absence of light yields 1.2cm.
(i) C6H5-CH2Cl (benzyl chloride) (ii) o-chlorotoluene (iii) p-chlorotoluene (iv) mixture of (ii) and (iii)
Correct option: (iv) mixture of (ii) and (iii).
Concept used.Fe/FeCl3 in the dark drives ring
chlorination via electrophilic substitution. The -CH3 group
on toluene is an o/p-director (activating, +I and
hyperconjugation), so both o- and p-chlorotoluene form.
Generate Cl+ via FeCl3.
Methyl on toluene donates into the ring; positions o and
p to it are the most nucleophilic.
Both isomers form; p tends to be the major because of
less steric clash with the methyl, but both are produced.
Both o- and p-chlorotoluene; option (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Director-first thinking. ``Lewis acid + dark + ring with
CH3'' is the signature of EAS at o/p. Any answer that
gives a single isomer is wrong –- both o and p form.
Concept used. Activating o/p-directors give a mixture
(∼ 60% p, ∼ 35% o, <5% m in practice). p is the
major because the σ-complex from o-attack carries some
steric strain between CH3 and the incoming Cl.
Confirm ring substitution (Lewis acid catalyst, dark).
Confirm o/p directing of CH3.
Pick (iv).
Option (iv).
Q 6.21
Chloromethane on treatment with excess of ammonia yields mainly
(i) N,N-dimethylmethanamine ((CH3)3N) (ii) N-methylmethanamine (CH3NHCH3) (iii) methanamine (CH3NH2) (iv) mixture containing all these in equal proportion
Correct option: (iii) methanamine (CH3NH2).
Concept used. Ammonolysis of an alkyl halide proceeds via
SN2. The first product, R-NH2, is itself a
nucleophile and can react further (polyalkylation). Excess
ammonia keeps the ratio [NH3]/[R-NH2] very high, so
NH3 wins the competition for R-X and the primary
amine is the dominant product.
Without excess NH3, CH3NH2 would react further
to give (CH3)2NH, (CH3)3N, and eventually
(CH3)4N+Cl-.
With excess NH3, the first product wins
⇒ methanamine.
CH3NH2 is the major product with excess NH3; option (iii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Statistical-control angle. The trick is the word
``excess''. The reaction is a chain of SN2 steps; each
step's rate is proportional to the concentration of the current
amine. Drowning the reaction in NH3 makes the first step
dominate by sheer numerical preponderance.
Concept used. Polyalkylation is a well-known nuisance of
Hofmann ammonolysis. Industrial workarounds: use excess NH3
(as here), or use KCN + LiAlH4 (Mendius), or use the
Gabriel-phthalimide synthesis to install primary amines cleanly.
Recognise SN2 on CH3Cl by NH3.
Excess NH3⇒ first product (1∘ amine) dominates.
Option (iii) methanamine.
Q 6.22
Molecules whose mirror image is non-superimposable over them are known as chiral. Which of the following molecules is chiral in nature?
(i) 2-Bromobutane (ii) 1-Bromobutane (iii) 2-Bromopropane (iv) 2-Bromopropan-2-ol
Correct option: (i) 2-Bromobutane.
Concept used. A molecule is chiral if it contains at least
one sp3 carbon bonded to four different groups (no plane
of symmetry).
(i) CH3-C*HBr-CH2CH3: four groups
(H, Br, CH3, C2H5) all different ⇒
chiral.
(ii) Br-CH2-CH2-CH2-CH3: C-Br carbon has
two Hs ⇒ not chiral.
(iii) (CH3)2CH-Br: C-Br carbon has two
identical methyls ⇒ not chiral.
(iv) (CH3)2C(OH)(Br)? Actually the name
``2-bromopropan-2-ol'' is CH3-C(Br)(OH)-CH3 –- two
methyl groups make C2 symmetric ⇒ not chiral.
Only 2-bromobutane is chiral; option (i).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Four-different rule applied four times. Test each option's
C-Br carbon for four distinct substituents.
Concept used. Chirality at a single tetrahedral centre
requires four different groups; identical alkyl substituents
(CH3,CH3) immediately disqualify.
Locate C-Br.
Count distinct substituents.
Only (i) clears the test.
Option (i).
Q 6.23
Reaction of C6H5-CH2Br with aqueous sodium hydroxide follows 1.2cm.
(i) SN1 mechanism (ii) SN2 mechanism (iii) Any of the above two depending upon temperature (iv) Saytzeff rule
Correct option: (i)SN1 mechanism.
Concept used. Benzyl halides ionise readily because the
intermediate benzyl cation C6H5-C+H2 is heavily
resonance-stabilised by the aromatic ring (three additional
resonance structures spread the positive charge onto o and p
ring carbons). Even though it is formally primary, it follows
SN1 kinetics in polar protic solvents like aq. NaOH.
Substrate: benzyl bromide C6H5CH2Br.
Solvent: aq. NaOH (polar protic, ionising).
Ionise to C6H5-CH2+ (resonance-stabilised) +Br-.
OH- traps the cation →C6H5CH2OH
(benzyl alcohol).
SN1 via resonance-stabilised benzyl cation; option (i).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Cation-resonance angle. The benzyl cation has four
resonance structures (positive charge on the CH2 carbon and
on each of o, o', p). That stabilisation drops the SN1
activation energy below the SN2 alternative for benzyl
halides in protic solvent.
Concept used. Polarity of solvent + cation stability
together select the dominant mechanism. Aqueous medium solvates
both the cation and the halide; ionisation is energetically downhill;
SN1 kinetics result.
Sketch all benzyl-cation resonance forms.
Conclude that ionisation is feasible at room temperature.
SN1 wins.
Option (i)SN1.
Q 6.24
Which of the carbon atoms present in the molecule given below are asymmetric? HOOC-Ca(O)-CbH(OH)-CcH(OH)-CdHO [2pt]
(i) a, b, c, d (ii) b, c (iii) a, d (iv) a, b, c
Correct option: (ii)b, c.
Concept used. A carbon is asymmetric only if it bears four
different groups. The carboxyl carbon (a) is a C=O
(only three substituents at sp2), and the aldehydic carbon (d)
is likewise sp2 (C(=O)H). Only b and c are sp3 with
four different groups.
a: C=O with HO- and -CH(OH)-. Only
three substituents (sp2), not asymmetric.
b: CH(OH) with neighbours COOH, CHOH-CHO, OH, H
–- four different groups. Asymmetric.
c: CH(OH) with neighbours CH(OH)-COOH, CHO, OH, H
–- four different groups. Asymmetric.
d: CHO (sp2), only three substituents.
Asymmetric carbons: b and c; option (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Hybridisation filter. An asymmetric carbon must be sp3
with four different substituents. Any sp2 carbon (carbonyl,
aldehyde, alkene) is automatically excluded because it has only
three substituents and is trigonal planar.
Concept used. The classic 4-carbon sugar acid this molecule
resembles (tartaric/threonic family) carries two consecutive
CHOH chiral centres flanked by carboxyl and aldehyde groups.
2n stereoisomers = 4 here (n=2), giving the (2R,3R), (2S,3S),
(2R,3S), and (2S,3R) diastereomers.
Strike off a and d (sp2).
Confirm b and c are sp3 with four different groups.
Option (ii)b, c.
Q 6.25
Which of the following compounds will give racemic mixture on nucleophilic substitution by OH- ion?
(a) CH3-CHBr-C2H5 (b) CH3-CBr(C2H5)(CH3) (c) CH3-CH(C2H5)-CH2Br [2pt]
(i) (a) (ii) (a), (b), (c) (iii) (b), (c) (iv) (a), (c)
Correct option: (i) (a) only.
Concept used. A racemic mixture forms when nucleophilic
substitution destroys chirality at the substrate carbon by going
through a planar carbocation (or by attacking a chiral SN1
substrate where the cation goes flat). Therefore we need:
1pt
the substrate must itself be chiral (so racemisation has
meaning); and
the mechanism must produce a planar intermediate
(here, the carbocation in SN1, or symmetric
SN2 on a chiral C if applicable).
(a) CH3-C*HBr-C2H5 (= 2-bromobutane): chiral
(H, Br, CH3, C2H5 all different). Secondary halide
⇒ on SN1 via planar
CH3-C+H-C2H5 gives 50:50 enantiomers
⇒ racemic.
(b) Tertiary, but CBr(CH3)2C2H5 has two
identical CH3 on the cation centre ⇒not chiral to start with; substitution gives a
non-chiral product. No racemisation.
(c) Primary R-CH2Br: not chiral at C-Br.
Substitution gives a non-chiral alcohol; no racemisation.
Only (a) gives a racemic mixture; option (i).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Pre-condition check. Two screens must be passed: (1)
substrate carbon is chiral; (2) mechanism passes through a planar
intermediate. (b) fails (1); (c) fails (1); only (a) passes both.
Concept used. Racemisation requires the chiral information
on the substrate to be destroyed during the substitution. The most
common route is SN1 on a chiral substrate: the cation is
flat, the nucleophile attacks either face equally, and the product is
a 50:50 mixture of enantiomers.
Test each substrate for chirality at C-Br.
Test each for SN1 feasibility.
Only (a) clears both screens.
Option (i).
Q 6.26
Arrange the following compounds in increasing order of rate of reaction towards nucleophilic substitution:
(a) chlorobenzene (b) o-nitrochlorobenzene (c) m-nitrochlorobenzene [2pt]
(i) (a) < (b) < (c) (ii) (c) < (b) < (a)
(iii) (a) < (c) < (b) (iv) (c) < (a) < (b)
Correct option: (iii)(a) < (c) < (b).
Concept used. The -NO2 group activates an aryl halide
toward SNAronly when it sits at o or p to the
halogen. From the m-position the negative charge of the
Meisenheimer adduct cannot delocalise onto the nitro oxygens
(resonance only touches o and p carbons).
(a) chlorobenzene: no activation, baseline (very slow).
(c) m-nitro: NO2 is m-located, no resonance
stabilisation of Meisenheimer; only mild -I activation.
(b) o-nitro: NO2 is o-located, full resonance
stabilisation ⇒ much faster.
Order: a < c < b; option (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Resonance-position angle. The acid test for SNAr
activation is whether you can draw a resonance structure that puts
the negative charge on a NO2 oxygen. From o and p you
can; from m you cannot.
Concept used. The Meisenheimer intermediate is a
delocalised cyclohexadienide anion; NO2 at o or p
intercepts the negative charge on its formal N=O oxygen,
slashing the energy by tens of kJ/mol. m-NO2 misses by one
ring atom.
Draw Meisenheimer for o-nitro → confirm (-) on NO2 O.
Draw Meisenheimer for m-nitro →(-) stuck on ring C.
Rate: o > m > none. Increasing order: a < c < b.
Option (iii)a < c < b.
Q 6.27
Arrange the following compounds in increasing order of rate of reaction towards nucleophilic substitution:
(a) chlorobenzene (b) o-chlorotoluene (c) m-chlorotoluene [2pt]
(i) (a)<(b)<(c) (ii) (a)<(c)<(b)
(iii) (c)<(b)<(a) (iv) (b)<(c)<(a)
Correct option: (iv)(b)<(c)<(a).
Concept used. A -CH3 group is electron-donating
(+I, hyperconjugation), so it destabilises the Meisenheimer
intermediate by adding electron density to a ring that is already
trying to accommodate a negative charge. The closer the CH3
is to the reaction site (i.e. at o), the worse the destabilisation.
Therefore reactivity drops with CH3 proximity.
(a) chlorobenzene: baseline, no CH3 on the ring.
(c) m-chlorotoluene: CH3 at the m-position, mild
+I slows it slightly.
(b) o-chlorotoluene: CH3 right next to Cl;
hyperconjugation directly destabilises the Meisenheimer
carbon adjacent to CH3⇒ slowest.
Order: b < c < a; option (iv).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Electron-donor angle. For SNAr, donors hurt and
acceptors help. The o-isomer is hurt most, the unsubstituted
parent is the baseline, the m-isomer is intermediate.
Concept used.CH3 is a weak +I/+H donor that
destabilises a Meisenheimer intermediate. The destabilisation falls
off with distance from the reacting carbon: o > m > p (and
notice the slight quirk that p is sometimes also strongly
destabilising for the same reason).
Rank donor strength to the carbanionic ring carbon: o > m > none.
Reactivity (rate) opposite: none > m > o.
Increasing order: b < c < a.
Option (iv)b < c < a.
Q 6.28
Arrange the following compounds in increasing order of rate of reaction towards nucleophilic substitution:
(a) chlorobenzene (b) p-nitrochlorobenzene (c) 2,4,6-trinitrochlorobenzene [2pt]
(i) (c)<(b)<(a) (ii) (b)<(c)<(a)
(iii) (a)<(c)<(b) (iv) (a)<(b)<(c)
Correct option: (iv)(a)<(b)<(c).
Concept used. Each additional o/p-NO2 adds another
resonance sink for the Meisenheimer carbanion. The activation effect
is roughly multiplicative; three suitably placed NO2 groups
boost the rate by ∼ 1012 over chlorobenzene.
(a) chlorobenzene: baseline.
(b) p-nitrochlorobenzene: one resonance sink at p;
reacts with aq. NaOH at ∼ 100∘C.
(c) picryl chloride (2,4,6-trinitrochlorobenzene): three
NO2 (one at p, two at o); reacts with H2O
at room temperature.
Order: a < b < c; option (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Stacking-acceptor angle. The Meisenheimer intermediate's
negative charge is delocalised onto o/p-NO2 oxygens.
Adding a second NO2 doubles the number of delocalisation
sinks; adding a third triples it. Activation barriers fall in step.
Concept used. Multiple -M groups operate
near-independently in stabilising the same intermediate, giving a
near-product relationship in rate constants. Picryl chloride is the
canonical example of multiple-nitro activation.
Count o/p-NO2 groups.
Apply: rate scales steeply with that count.
Order: a (0 nitros) <b (1 nitro) <c (3 nitros).
Option (iv).
Q 6.29
Arrange the following compounds in increasing order of rate of reaction towards nucleophilic substitution:
(a) chlorobenzene (b) p-methylchlorobenzene (p-chlorotoluene)
(c) 2,4-dimethylchlorobenzene [2pt]
(i) (a)<(b)<(c) (ii) (b)<(a)<(c)
(iii) (c)<(b)<(a) (iv) (a)<(c)<(b)
Correct option: (iii)(c)<(b)<(a).
Concept used. Methyl groups are electron donors (+I /
hyperconjugation); they deactivate an aryl halide toward
SNAr by destabilising the negatively charged Meisenheimer
intermediate. Two methyl groups deactivate roughly twice as much as
one.
(a) chlorobenzene: baseline (least deactivated).
(b) p-chlorotoluene: one CH3, slower than (a).
(c) 2,4-dimethylchlorobenzene: two CH3 (both
o/p), even slower.
Order: c < b < a; option (iii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Donor-stacking angle. Each additional CH3 at o/p
amplifies the destabilisation of the Meisenheimer carbanion. Order
of reactivity inverts the order of CH3 count.
Concept used. Electron-donating groups on a ring already
bearing a negative charge are destabilising. The effect adds: more
donors ⇒ slower SNAr.
Count o/p-CH3 groups.
Reactivity: 0 (a) > 1 (b) > 2 (c). Increasing rate: c < b < a.
Option (iii).
Q 6.30
Which is the correct increasing order of boiling points of the following compounds?
1-Iodobutane, 1-Bromobutane, 1-Chlorobutane, Butane. [2pt]
(i) Butane < 1-Chlorobutane < 1-Bromobutane < 1-Iodobutane
(ii) 1-Iodobutane < 1-Bromobutane < 1-Chlorobutane < Butane
(iii) Butane < 1-Iodobutane < 1-Bromobutane < 1-Chlorobutane
(iv) Butane < 1-Chlorobutane < 1-Iodobutane < 1-Bromobutane
Correct option: (i) Butane < 1-Cl < 1-Br < 1-I.
Concept used. For the same alkyl skeleton, alkyl halide
boiling points rise with halogen mass (heavier halide ⇒
more electrons ⇒ more polarisable ⇒ stronger
dispersion forces). Butane itself is a hydrocarbon (no halogen),
so it boils lowest.
Butane: ∼ 273 K (just above water's freezing point).
1-Chlorobutane: ∼ 351 K.
1-Bromobutane: ∼ 374 K.
1-Iodobutane: ∼ 403 K.
Order: butane < 1-Cl < 1-Br < 1-I; option (i).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mass-and-polarisability angle. Across the halogen group,
both molar mass and polarisability rise sharply
(F→I). Both push the boiling point up.
Concept used. Dispersion forces scale with polarisability;
boiling point scales with dispersion forces; therefore Tb scales
with halogen mass for the same alkyl chain.
Confirm same skeleton (butyl chain) across the three halides.
Add butane (no halide) at the bottom.
Order by halogen mass.
Option (i).
Q 6.31
Which is the correct increasing order of boiling points of the following compounds?
1-Bromoethane, 1-Bromopropane, 1-Bromobutane, Bromobenzene. [2pt]
(i) Bromobenzene < 1-Bromobutane < 1-Bromopropane < 1-Bromoethane
(ii) Bromobenzene < 1-Bromoethane < 1-Bromopropane < 1-Bromobutane
(iii) 1-Bromopropane < 1-Bromobutane < 1-Bromoethane < Bromobenzene
(iv) 1-Bromoethane < 1-Bromopropane < 1-Bromobutane < Bromobenzene
Concept used. Within the alkyl bromide series, boiling point
rises with chain length (more electrons, more vdW contact).
Bromobenzene, although same Br count, has the additional
six-carbon aromatic ring; its boiling point is the highest
(∼ 429 K) because of stronger dispersion forces from the
planar polarisable π-cloud.
Surface-area-plus-π angle. The aromatic ring of PhBr
adds substantial π-electron polarisability that no aliphatic
butyl chain can match, even chain-by-chain.
Concept used. Boiling point ∝ intermolecular
dispersion energy. Aromatic ring π-electrons are highly
polarisable; combined with the heavy Br, PhBr sits at the
top of this comparison.
Order alkyl bromides by chain length.
Compare PhBr's polarisability to the heaviest alkyl
bromide here (n-Bu-Br).
PhBr (∼ 429 K) tops 1-bromobutane (∼ 374 K).
Option (iv).
II. Multiple Choice Questions (Type-II)
Q 6.32
Which of the following compounds are gem-dihalides?
(i) Ethylidene chloride (ii) Ethylene dichloride (iii) Methylene chloride (iv) Benzyl chloride
Correct options: (i) and (iii).
Concept used. A gem-dihalide carries both halogens
on the same carbon atom. We need to identify all options
whose two halogens sit on one carbon.
(i) Ethylidene chloride: CH3-CHCl2. Both Cl
on C1⇒gem.
(ii) Ethylene dichloride: ClCH2-CH2Cl (= 1,2-DCE);
one Cl per carbon ⇒vic.
(iii) Methylene chloride: CH2Cl2. Both Cl on
the single carbon ⇒gem.
(iv) Benzyl chloride: C6H5-CH2Cl –- only one Cl,
not a dihalide at all.
gem-Dihalides: (i) and (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Connectivity-first angle. Draw out each substrate from
its trivial name and count how many halogens sit on each carbon.
gem means geminal, two halogens on the same carbon.
The trivial-name convention is consistent: ``-idene'' endings
indicate a divalent carbon (R-CHX2 or =CR2) and so
always carry two halogens on one carbon, while ``-ene''
substituents trail -CH2-CH2- and so put halogens on
adjacent carbons. Once you internalise that, you read gem
versus vic straight off the name.
Concept used. The class of a polyhalogenated compound
is set strictly by the connectivity of the halogen atoms, not
by their identity or count. We must therefore test each option's
structural formula for the ``same-carbon-or-not'' criterion.
Halogens identity (Cl, Br) is irrelevant to this
classification.
(i) Ethylidene chloride. ``Ethylidene'' denotes
a divalent CH3-CH= fragment, so both Cl
attach to the same (C1) carbon ⇒CH3-CHCl2, gem.
(ii) Ethylene dichloride. ``Ethylene'' denotes
the -CH2-CH2- fragment, so the chlorines sit one
each on C1 and C2⇒ClCH2-CH2Cl, vic.
(iii) Methylene chloride. ``Methylene'' denotes
the CH2 fragment, only one carbon available; both
Cl sit on that single carbon ⇒CH2Cl2, gem.
(iv) Benzyl chloride.C6H5-CH2Cl has
one halogen; it is a monohalide, not a dihalide,
so it cannot be classified gem or vic.
gem-Dihalides: (i) and (iii).
Q 6.33
Which of the following are secondary bromides?
(i) (CH3)2CHBr (ii) (CH3)3C-CH2Br (iii) CH3CH(Br)CH2CH3 (iv) (CH3)2CBrCH2CH3
Correct options: (i) and (iii).
Concept used. The class of an alkyl halide R-X is
set by the number of other carbon atoms attached to the
C-X carbon: 1∘ if one, 2∘ if two,
3∘ if three.
(i) (CH3)2CHBr: C-Br bears two CH3
groups ⇒2∘.
(ii) (CH3)3C-CH2Br: C-Br is a CH2
attached to one C (the C(CH3)3) ⇒1∘ (neopentyl bromide).
(iii) CH3-CH(Br)-CH2CH3: C-Br bears a CH3
and a CH2CH3⇒2∘.
(iv) (CH3)2C(Br)CH2CH3: C-Br bears three
carbons ⇒3∘.
Secondary bromides: (i) and (iii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Substituent-counting angle. Mask the Br atom
mentally and count the number of carbon neighbours of the
C-Br carbon. Exactly two ⇒ secondary. Hydrogens
do not count. This single-glance method scales to every alkyl
halide nomenclature question.
Concept used. Alkyl-halide classification depends only
on the connectivity at the halogen-bearing carbon. ``Secondary''
(2∘) means precisely two carbon substituents on the
C-X carbon. Watch out for spectator carbons: in
(CH3)3C-CH2Br the Br-bearing CH2 has only
one carbon neighbour (the C(CH3)3 block) even though
the molecule has four other carbons elsewhere. That makes it
neopentyl bromide, a primary halide that is famously sluggish
in SN2 due to neopentyl steric shielding.
(i) (CH3)2CHBr: mask Br; the CH
has two CH3 neighbours ⇒2∘.
(ii) (CH3)3C-CH2Br: mask Br; the
CH2 has one carbon neighbour (C(CH3)3)
⇒1∘ (neopentyl bromide).
(iii) CH3-CH(Br)-CH2CH3: mask Br;
the CH has CH3 and CH2CH3 neighbours
⇒2∘.
(iv) (CH3)2C(Br)CH2CH3: mask Br;
the central C has three carbon neighbours
(CH3,CH3,CH2CH3) ⇒3∘.
Therefore (i) and (iii) are the secondary bromides.
Secondary bromides: (i) and (iii).
Q 6.34
Alkyl halides are prepared from alcohols by treating with
(i) HCl + ZnCl2 (ii) Red P + Br2 (iii) H2SO4 + KI (iv) All the above
Correct options: (i) and (ii).
Concept used. Standard preparations of R-X from
R-OH:
HCl + ZnCl2 (Lucas reagent) for R-Cl.
Red P + Br2 (generates PBr3 in situ) for R-Br.
PI3 (red P + I2) for R-I; SOCl2
for clean R-Cl.
(i) Lucas: R-OH + HCl -> R-Cl + H2O with
ZnCl2 as catalyst. Standard prep.
(ii) Red P + Br2: 2P + 3Br2 -> 2PBr3, then
3R-OH + PBr3 -> 3R-Br + H3PO3.
(iii) H2SO4 + KI: conc.H2SO4 oxidises
I- to I2, so this fails for R-I.
Use H3PO4 + KI instead.
(i) and (ii) are correct prep methods; (iii) is wrong.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Acid-choice angle. The selection of a Brnsted acid
when making an alkyl halide is governed not just by acid strength
but by the redox compatibility between the acid and the
halide anion to be released. Hard rule: for Cl- and
Br-, H2SO4 is fine; for I-, you must use
H3PO4 or a non-oxidising substitute. Recognising this
single distinction kills option (iii) instantly.
Concept used. The standard preparations of R-X
from R-OH are three: (a) Lucas reagent
HCl/ZnCl2 (SN1 with ZnCl2 acting as a
Lewis acid that pre-activates -OH); (b) red P + Br2
which generates PBr3 in situ (2P + 3Br2 -> 2PBr3),
then 3R-OH + PBr3 -> 3R-Br + H3PO3; (c) red P + I2
to give PI3 for R-I. SOCl2 (R-OH + SOCl2
-> R-Cl + SO2 + HCl) is the cleanest because the by-products are
gaseous.
(i) HCl/ZnCl2 (Lucas). A textbook
SN1 on R-OH; ZnCl2 coordinates
the hydroxyl oxygen and assists ionisation. Valid.
(ii) Red P + Br2. Forms PBr3
in situ, which esterifies and substitutes the alcohol.
Valid for R-Br.
(iii) H2SO4 + KI. The acid oxidises the
I- liberated from KI to I2 and
SO2, so no HI is available to attack the
alcohol. Fails. Use H3PO4 + KI instead.
Hence (i) and (ii) are the workable methods.
(i) and (ii) are correct preparations; (iii) fails on redox grounds.
Q 6.35
Alkyl fluorides are synthesised by heating an alkyl chloride/bromide in presence of
(i) CaF2 (ii) CoF2 (iii) Hg2F2 (iv) NaF
Correct options: (ii) and (iii).
Concept used. The Swarts reaction converts
R-Br or R-Cl to R-F using a heavy-metal
fluoride that is sufficiently nucleophilic and thermally stable.
Standard reagents: AgF, Hg2F2, CoF2, SbF3.
NaF and CaF2 have very high lattice energies and
are essentially inert under typical conditions.
(i) CaF2: massive lattice energy, F- is not
available to attack R-X.
(ii) CoF2: classic Swarts reagent.
(iii) Hg2F2: mercurous fluoride is the original
Swarts reagent.
(iv) NaF: poorly nucleophilic in non-polar media;
not used for Swarts.
Swarts fluorides come from CoF2 or Hg2F2; (ii) & (iii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Reagent-recall angle. The Swarts reaction is mnemonically
``replace Cl/Br by F using a heavy-metal fluoride''. The two
NCERT-named candidates are Hg2F2 (mercurous fluoride, the
original 1892 Swarts reagent) and CoF2. Both options must
therefore be ticked. The two distractors (NaF, CaF2)
are the classical fluoride sources of ionic chemistry, but
neither delivers a nucleophilic fluoride to an organic substrate.
Concept used. The Swarts reaction is an SN2
halogen exchange in which a soft, polarisable metal fluoride
acts as an F- donor: R-Br + AgF -> R-F + AgBr.
For the reagent to work, the metal fluoride must (a) be soluble
or accessible enough in the reaction medium that F- is
liberated, and (b) form a thermodynamically stable metal halide
by-product (e.g. AgBr, HgBr2). Hard ionic
fluorides NaF, CaF2, LiF have lattice energies
of ∼ 920–2600 kJ/mol; that energy must be repaid before
the fluoride is mobile.
Test (i) CaF2. Lattice energy
∼ 2630 kJ/mol; effectively insoluble in organic
media. Inert. Reject.
Test (ii) CoF2. Listed in NCERT as a
Swarts reagent; the cobalt(II) centre is a soft cation
and the fluoride is sufficiently labile. Valid.
Test (iii) Hg2F2. The original Swarts
reagent; mercury(I) is a borderline-soft cation that
readily exchanges fluoride for chloride/bromide. Valid.
Test (iv) NaF. High lattice energy and
poor solubility in non-polar organic solvents; useless
for the Swarts protocol.
Swarts fluorides come from CoF2 or Hg2F2; options (ii) and (iii).
Q 6.36
Consider the SN2 reaction: HO- (a) + R-Cl (b) -> [HO⋯ C⋯ Cl]- (c) -> HO-R (d) + Cl- (e)
Which of the statements are correct about the above reaction?
(i) (a) and (e) both are nucleophiles (ii) In (c) carbon atom is sp3 hybridised (iii) In (c) carbon atom is sp2 hybridised (iv) (a) and (e) both are electrophiles
Correct options: (i) and (iii).
Concept used. In the SN2 transition state (c)
the substrate carbon is simultaneously bonded to incoming OH-
and outgoing Cl- on opposite faces (trigonal-bipyramidal),
so its bonding hybridisation is sp2 with the remaining p-orbital
shared between the two leaving/incoming groups. Both OH- (a)
and Cl- (e) are anions with lone pairs ⇒
nucleophiles.
(a) OH- has lone pairs ⇒ nucleophile.
(e) Cl- likewise a nucleophile (and the leaving group of the next round).
(c) The TS has 5 partial bonds to C ⇒sp2
with a p-lobe pointing along the OH⋯ C⋯ Cl axis.
Reject (iv): anions with lone pairs are never electrophiles.
Reject (ii): the TS is sp2 trigonal-bipyramidal, not sp3.
Correct: (i), (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
TS-geometry angle. The SN2 TS is the canonical
trigonal-bipyramidal structure: three retained substituents lie in a
plane around the central carbon, and the incoming/outgoing groups
occupy axial positions. That geometry forces the central C to use
three sp2 hybrids for the equatorial bonds; the remaining p
orbital is shared between Nu and LG.
Concept used. Hybridisation tracks σ-bond count and
geometry. The TS has three full σ-bonds in the equatorial
plane + two half-bonds along the axial direction ⇒sp2.
Identify nucleophiles in the equation: (a) and (e) both anions.
Identify TS geometry: trigonal-bipyramidal ⇒sp2 at C.
Match: (i), (iii).
Options (i) and (iii).
Q 6.37
For the reaction in Q32, which of the following statements are correct about this reaction?
(i) The given reaction follows SN2 mechanism (ii) (b) and (d) have opposite configuration (iii) (b) and (d) have same configuration (iv) The given reaction follows SN1 mechanism
Correct options: (i) and (ii).
Concept used. The reaction shows a single concerted step
with simultaneous bond-making and bond-breaking, with a TS
containing both Nu and LG; this is the defining signature of
SN2. Back-side attack produces Walden inversion of the
configuration at the substrate carbon.
Concerted single-step TS ⇒SN2.
Back-side attack of OH- from the face opposite to
Cl inverts the spatial arrangement at C.
Therefore (b) and (d) have opposite configurations.
Correct: (i), (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Walden-inversion angle. Whenever a single-step bimolecular
substitution is shown with explicit back-side attack, the product
must be the inverted stereoisomer of the substrate.
Concept used.SN2 stereo outcome: 100%
inversion of the substrate's relative configuration. If the
substrate is R, the product is S (assuming no priority change
of the substituents).
Recognise concerted TS ⇒SN2.
Recognise back-side attack ⇒ inversion.
Options (i), (ii).
Q 6.38
Which of the following statements are correct about the reaction intermediate (c) in Q32?
(i) Intermediate (c) is unstable because in this carbon is attached to 5 atoms (ii) Intermediate (c) is unstable because carbon atom is sp2 hybridised (iii) Intermediate (c) is stable because carbon atom is sp2 hybridised (iv) Intermediate (c) is less stable than the reactant (b)
Correct options: (i) and (iv).
Concept used. The species (c) is in fact a transition
state, not a true intermediate; nevertheless the question treats it
as an intermediate. The five partial bonds violate the normal
octet/tetravalency of carbon, making it a very high-energy species.
It is also significantly higher in energy than the starting reactant
(b), as required for any reaction passing over a barrier.
Energy of TS > energy of starting reactant (it sits at the activation peak).
Reject (ii): sp2 hybridisation itself is not destabilising;
it's the over-coordination that destabilises.
Reject (iii): a TS by definition is unstable, not stable.
Correct: (i), (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Energetics angle. Any species that lies above the
reactants on the reaction coordinate is, by definition, less stable
than the reactants. The TS of SN2 sits at the peak; it
features a hypervalent (pentacoordinate) carbon.
Concept used. Transition-state theory: the TS is the
maximum of the reaction-coordinate energy profile; activation
energy Ea = ETS - Ereactants > 0.
Identify (c) as the TS.
Apply transition-state theory: TS is unstable, energy > reactants.
Pentacoordinate C ⇒ violates normal valence.
Options (i), (iv).
Q 6.39
Consider the reaction: HO- (a) + Et-CHCl-Me (b) -> [HO⋯ C⋯ Cl]- -> Et-CH(OH)-Me (c) + Cl- (d).
Which of the following statements are correct about the mechanism of this reaction?
(i) A carbocation will be formed as an intermediate in the reaction (ii) OH- will attach the substrate (b) from one side and Cl- will leave it simultaneously from other side (iii) An unstable intermediate will be formed in which OH- and Cl- will be attached by weak bonds (iv) Reaction proceeds through SN1 mechanism
Correct options: (ii) and (iii).
Concept used. The substrate is a secondary alkyl
halide (CH3-CHCl-C2H5). With OH- (strong nucleophile)
in aqueous medium, it undergoes SN2 –- a single concerted
step with back-side attack. No carbocation intermediate forms; the
TS features partial bonds from OH- and to Cl-.
(ii) is the textbook description of SN2 back-side attack.
(iii) describes the trigonal-bipyramidal TS where both
partial bonds are weak (long, partial).
Reject (i) and (iv): no carbocation, no SN1.
Correct: (ii), (iii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Mechanism-classification angle. Secondary halide + strong
nucleophile + aqueous medium ⇒SN2 (with minor
SN1 if substrate is benzyl/allyl, which this isn't).
Concept used. The fork in the road: SN1 goes
through a carbocation; SN2 goes through a single TS.
Choice is set by substrate class, nucleophile strength, and solvent.
Tag substrate class: secondary, mildly activated.
Tag conditions: aqueous, strong OH-.
Mechanism: SN2.
Options (ii), (iii).
Q 6.40
Which of the following statements are correct about the kinetics of the reaction in Q35?
(i) The rate of reaction depends on the concentration of only (b) (ii) The rate of reaction depends on concentration of both (a) and (b) (iii) Molecularity of reaction is one (iv) Molecularity of reaction is two
Correct options: (ii) and (iv).
Concept used.SN2 is bimolecular: the rate law
is rate = k[R-X][Nu-], and the molecularity
(number of species in the rate-determining step) is two.
Write rate law: rate = k[OH-][R-Cl].
Both concentrations enter; molecularity =2.
Rejection of (i) and (iii): those describe SN1.
Correct: (ii), (iv).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Rate-law angle. The rate-determining step of
SN2is the bond-making/bond-breaking step; both
the substrate and the nucleophile are reactants in that step, so
both appear in the rate law.
Concept used. Molecularity counts the participants in the
rate-determining step; rate law also reflects them. For
SN2, two participants ⇒ molecularity =2,
order =2.
Establish SN2.
Write rate law.
Pick (ii) and (iv).
Options (ii), (iv).
Q 6.41
Haloalkanes contain halogen atom(s) attached to the sp3 hybridised carbon atom of an alkyl group. Identify haloalkane from the following compounds.
(i) 2-Bromopentane (ii) Vinyl chloride (chloroethene) (iii) 2-Chloroacetophenone (iv) Trichloromethane
Correct options: (i) and (iv).
Concept used. A haloalkane has the halogen on an sp3
carbon of an alkyl chain. (ii) chloroethene has Cl on sp2C=C carbon (vinyl halide); (iii) chloroacetophenone has
Cl on the sp3CH2 next to C=O, but this is
strictly α-haloketone –- some count it; NCERT keys it as
correct. Re-check.
(i) CH3-CHBr-CH2CH2CH3: Br on sp3 C ⇒ haloalkane.
(ii) CH2=CHCl: Cl on sp2 C ⇒ vinyl halide, not haloalkane.
(iii) C6H5-CO-CH2Cl: Cl on sp3CH2;
this is an α-haloketone, not a pure haloalkane.
(iv) CHCl3: Cl on a sole sp3 C; haloalkane.
Correct: (i), (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Hybridisation filter. ``Haloalkane'' requires the halogen
on an sp3 carbon and that carbon to belong to an alkyl skeleton.
Eliminate sp2 (vinyl, aryl) and non-alkyl (acyl, α-keto)
options.
Concept used. Classification: sp3 C alkyl chain → haloalkane. sp2 C of C=C→ vinyl halide. sp2 C of C=O→ acyl halide.
Aryl ring → aryl halide.
Inspect each option's C-X hybridisation.
Pick those with sp3 alkyl skeleton.
Options (i), (iv).
Q 6.42
Ethylene chloride and ethylidene chloride are isomers. Identify the correct statements.
(i) Both the compounds form same product on treatment with alcoholic KOH (ii) Both the compounds form same product on treatment with aq. NaOH (iii) Both the compounds form same product on reduction (iv) Both the compounds are optically active
Correct options: (i) and (iii).
Concept used. Ethylene chloride =ClCH2-CH2Cl
(vic), ethylidene chloride =CH3-CHCl2 (gem).
Both undergo double dehydrohalogenation with alc. KOH to
give the same acetylene (after loss of 2 HCl), and both give
ethane on reduction (loss of both Cl replaced by H).
(i) Alc. KOH on ClCH2-CH2Cl: 2 HCl elimination →HC#CH (acetylene). Alc. KOH on CH3-CHCl2:
also loses 2 HCl →HC#CH. Same product.
(iii) Reduction (Zn/HCl, LiAlH4): both lose 2 Cl→CH3CH3 (ethane). Same product.
(iv) Neither compound has a chiral centre ⇒
not optically active.
Correct: (i), (iii).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Isomer-products angle. Two isomeric dihalides give the same
product whenever the operation (a) removes the halogens (reduction)
or (b) creates the same unsaturated framework (double
dehydrohalogenation). Operations that differentiate them (aqueous
NaOH gives diol vs gem-diol → aldehyde) yield different products.
Concept used. Alc. KOH does E2, doubly here
⇒ acetylene from any 1,1- or 1,2-dichloroethane. Aq.
NaOH does SN, so a vic-dihalide → diol, gem-dihalide
→ gem-diol → aldehyde/ketone (it loses water).
Predict products of each operation for each isomer.
Compare: same in (i), (iii); different in (ii); irrelevant in (iv).
Options (i), (iii).
Q 6.43
Which of the following compounds can be classified as aryl halides?
(i) p-ClC6H4-CH2CH(CH3)2
(ii) p-CH3CHCl(C6H4)CH2CH3
(iii) o-BrH2C-C6H4-CH(CH3)CH2CH3
(iv) C6H5-Cl
Correct options: (i) and (iv).
Concept used. An aryl halide carries the halogen on a ring
carbon. In (i) the Cl is on the ring (the substituent
CH2CH(CH3)2 is an alkyl tail attached at the para position
of a chlorobenzene); in (iv) C6H5-Cl is chlorobenzene
itself. In (ii) and (iii) the halogen sits on a side-chain
CH2/CHCl carbon, not on the ring ⇒ benzyl-type
alkyl halides.
(i) p-(Cl)C6H4-R: Cl on the ring ⇒ aryl halide.
(ii) The Cl is on the benzyl CHCl-CH3 side
chain ⇒ alkyl (benzyl) halide.
(iii) The Br is on a CH2 attached to the ring
⇒ benzyl (alkyl) halide.
(iv) C6H5-Cl: Cl on the ring ⇒ aryl halide.
Aryl halides: (i) and (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Halogen-location angle. ``Where is the halogen?'' is the
single test. On a ring carbon ⇒ aryl halide. On any side
chain sp3 carbon ⇒ alkyl halide (benzyl if next to
ring).
Concept used. Aryl halides are characterised not by the
presence of a ring but by the C-X being part of the ring.
Benzyl halides (C6H5-CH2-X) are alkyl halides whose alkyl
group happens to be benzyl.
For each option, locate X and check whether its
carbon is part of the aromatic ring.
Pick (i) and (iv).
Options (i), (iv).
III. Short Answer Type
Q 6.44
Aryl chlorides and bromides can be easily prepared by electrophilic substitution of arenes with chlorine and bromine respectively in the presence of Lewis acid catalysts. But why does preparation of aryl iodides require presence of an oxidising agent?
Concept used. Iodination of an arene is intrinsically
reversible: Ar-H + I2 <=> Ar-I + HI. The by-product
HI is a strong reducing agent; it reduces Ar-I back
to Ar-H and itself becomes I2. To pull the
equilibrium forward we must destroyHI as it forms.
Add an oxidising agent (typically HIO3, HNO3
or Hg(OAc)2) to the reaction mixture.
The oxidant converts the newly formed HI back to
I2: 5HI + HIO3 -> 3I2 + 3H2O.
Removing HI drives the equilibrium toward Ar-I
(Le Chatelier), so iodination now proceeds to completion.
The oxidant is therefore not the iodinating species; it is a
driving-force regulator that prevents back-reaction.
Iodination is reversible; oxidant removes HI to push the equilibrium toward Ar-I.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Equilibrium-first angle. The single insight that explains
the whole question is reversibility. While chlorination and
bromination of arenes are practically irreversible
(Δ G ≪ 0 because of strong C-Cl and C-Br
bonds plus stable HCl/HBr by-products), iodination
sits at a much smaller, even positiveΔ G. The
Ar-I bond is weak (∼ 240 kJ/mol) and the HI
by-product is itself a powerful reducing agent that can reverse
the substitution. So an oxidant is required not to make the
forward step faster, but to prevent the reverse step.
Concept used. The position of equilibrium in a
reversible EAS depends on bond strengths and on the stability
of the by-product acid. We use Le Chatelier's principle: remove
HI as fast as it forms, and the equilibrium
Ar-H + I2 <=> Ar-I + HI slides to the right. Common
oxidants used to achieve this are HIO3, HNO3, and
Hg(OAc)2; each converts HI either back to
I2 (more iodinating agent!) or to harmless species like
H2O and metallic iodides.
Diagnose the issue. The forward step
Ar-H + I2 -> Ar-I + HI is mildly endothermic;
without intervention, HI accumulates and reduces
Ar-I back to Ar-H.
Add the oxidant. A typical choice is iodic
acid: 5HI + HIO3 -> 3I2 + 3H2O. HI is
destroyed and additional I2 is regenerated for
more substitution.
Result. The equilibrium is permanently
displaced toward Ar-I, so iodination now proceeds
cleanly to completion. The oxidant itself does not
deliver I+ –- it is a thermodynamic regulator.
Oxidant oxidises HI back to I2/water, pulling the reversible equilibrium toward Ar-I.
Q 6.45
Out of o- and p-dibromobenzene which one has higher melting point and why?
Concept used. Melting point of an isomer is set by how
efficiently the molecules pack in the crystal lattice.
A more symmetric molecule packs tighter and needs more thermal
energy to break the lattice ⇒ higher m.p.
Draw o-dibromobenzene and p-dibromobenzene. The p-
isomer is centrosymmetric: a C2 axis perpendicular
to the ring and a centre of inversion at the ring centre.
The o- isomer has lower symmetry (mirror plane only),
so it stacks less efficiently.
The p-isomer wins by ∼ 80∘C because
of better crystal packing.
p-Dibromobenzene has a higher melting point (∼ 87∘C) than the o-isomer (∼ 7∘C) because its higher symmetry allows tighter crystal packing.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Packing-first angle. The melting point of a molecular
solid is determined by how efficiently molecules tile the
crystal lattice and how strong the van der Waals contacts
between them are. Higher symmetry means a more compact, more
regular unit cell, which gives a higher lattice energy and
therefore a higher melting point. For o- vs p-dibromobenzene
the result is dramatic –- a difference of nearly 80∘C
purely from crystal packing.
Concept used. Symmetric isomers carry an inversion
centre or a higher-order proper-rotation axis, so identical
molecules align in alternating layers and bury more van der Waals
contact area per unit volume. Asymmetric isomers leave dipole
fragments mismatched in the lattice; packing efficiency suffers.
This is why aromatic textbooks routinely report p ≫ m ≈ o
for melting points of disubstituted benzenes (boiling points,
which depend on dipole moment, do not show the same trend).
Symmetry analysis.p-Dibromobenzene possesses a C2 axis perpendicular
to the ring and a centre of inversion at the ring centre;
o-dibromobenzene has only a C2v mirror plane and
no inversion centre.
Packing consequence. The centro-symmetric p-
isomer stacks in a brick-like motif with maximised
π–π and BrMATH0Br contacts;
the o- isomer carries its two Br on the same
side, so packing leaves voids.
Quantitative result. Observed m.p. values:
o-dibromobenzene ≈ 7∘C;
p-dibromobenzene ≈ 87∘C. The p- isomer
wins by ∼ 80∘C.
p-Dibromobenzene melts at ≈ 87∘C vs ≈ 7∘C for the o- isomer, because its centro-symmetric packing is far more efficient.
Q 6.46
Write the structure of the compound whose IUPAC name is 1-Bromo-4-sec-butyl-2-methylbenzene. Use a TikZ sketch.
Concept used. Number the benzene ring so that the principal
substituent (Br) gets the lowest locant (1), then the
substituents in alphabetical order get the next-lowest locants
consistent with that choice. ``sec-butyl'' is
CH3-CH(C2H5)-, attached at its C2.
Place Br on C1.
Place -CH3 on C2.
Place sec-butyl (-CH(CH3)CH2CH3) on C4.
!%
[See diagram in the PDF version]
%
Structure: Br on C1, CH3 on C2, CH(CH3)CH2CH3 on C4 of benzene.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Locant-first angle. For a polysubstituted arene, lock
the principal substituent at locant 1 first, then walk around
the ring in whichever direction minimises the next locant. The
final locant set must obey the ``lowest set of locants'' rule.
Once those positions are fixed, attach each substituent at its
locant exactly as named.
Concept used. For a benzene with halogen and alkyl
substituents, the halogen (here Br) is the principal
substituent because there is no carboxylic acid, sulphonic acid,
nitro, etc. to outrank it. The sec-butyl group means
-CH(CH3)CH2CH3, i.e. a butyl that is attached through
its C2 carbon (hence the ``sec'' = secondary attachment).
Drawing requires placing each substituent on the correct ring
carbon and showing the attachment point of the side chain.
Anchor Br at C1. The principal
substituent takes the lowest locant.
Choose direction. Numbering clockwise:
C1(Br), C2(CH3), C3, C4,
C5, C6 –- this gives the methyl the
lowest locant 2 (alternative anticlockwise would put
methyl at locant 6, far higher).
Attach sec-butyl at C4. The
substituent is -CH(CH3)CH2CH3; the bond from the
ring goes to the central CH of the butyl chain.
Verify locant set.1, 2, 4 is lower than
the alternative 1, 4, 6, so the chosen direction
is correct.
Br at C1, CH3 at C2, sec-butyl at C4 of benzene –- as drawn in the structure above.
Q 6.47
Which of the following compounds would undergo SN1 reaction faster and why?
(A) CH2=CH-CH2-Cl (allyl chloride)
(B) CH3-CH2-CH2-Cl (n-propyl chloride)
Concept used.SN1 rate ∝ carbocation
stability. Allyl chloride ionises to the allyl cationCH2=CH-CH2+, which is resonance-stabilised:
CH2=CH-CH2+ <-> +CH2-CH=CH2. n-Propyl chloride would have to ionise to a primary
CH3CH2CH2+ with no resonance stabilisation.
Draw both cations; note the allyl cation is delocalised
over two carbons, giving it ∼ 60 kJ/mol of extra
resonance stabilisation.
The propyl cation is a primary ion –- very high energy.
Therefore CH2=CH-CH2-Cl ionises far more readily
⇒SN1 rate is much higher for (A).
(A) Allyl chloride is faster in SN1 because the allyl cation is resonance-stabilised.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Resonance-first angle. Whenever a leaving group sits
next to a π-system (allyl, benzyl, α to a carbonyl,
etc.), the cation that would form upon ionisation is no longer
the formal class you'd guess from substitution count –- it is
resonance-stabilised over the adjacent multiple bonds.
For allyl chloride this is decisive: the cation is delocalised
across two carbons, giving it stability comparable to a secondary
ion and making SN1 orders of magnitude faster than
for n-propyl chloride.
Concept used.SN1 rate depends on the
stability of the carbocation intermediate, which in turn depends
on how delocalised its positive charge is. The allyl cation
CH2=CH-CH2+ is symmetric across two carbons; the two
resonance structures (shown in the diagram above) contribute
equally, and the bond order between C1 and C2
becomes exactly 1.5. By Hammond's postulate the activation
energy for forming the allyl cation is correspondingly lower
than for forming the n-propyl cation (a localised primary
ion), so allyl chloride beats n-propyl chloride in any
SN1 comparison.
Draw both potential cations. Allyl:
CH2=CH-CH2+ <-> +CH2-CH=CH2, two equivalent
resonance forms (sketch above). n-Propyl:
CH3CH2CH2+, no resonance partners.
Estimate stabilisation. The allyl cation enjoys
∼ 60 kJ/mol of extra resonance stabilisation
relative to a localised primary cation; the n-propyl
cation has none.
Rank. Lower-energy cation ⇒ lower
Ea for ionisation ⇒ faster SN1.
Allyl chloride wins.
(A) Allyl chloride ionises faster because the allyl cation is resonance-stabilised over two carbons.
Q 6.48
Allyl chloride is hydrolysed more readily than n-propyl chloride. Why?
Concept used. Hydrolysis R-Cl + H2O -> R-OH + HCl
typically proceeds by SN1 (or SN2 for very
unhindered primaries). For allyl chloride the SN1 rate
dominates because the intermediate allyl cation is
resonance-delocalised.
Ionise allyl chloride: CH2=CH-CH2-Cl -> CH2=CH-CH2+
<-> +CH2-CH=CH2. The charge is shared by two carbons.
Ionise n-propyl chloride: CH3CH2CH2-Cl -> CH3CH2CH2+,
a primary, non-stabilised cation. Much slower.
Water attacks each cation to give the alcohol; the rate is
controlled by the first (ionisation) step.
Hence allyl chloride hydrolyses substantially faster than the
n-propyl analogue.
Allyl chloride hydrolyses faster because the allyl cation is resonance-stabilised over two carbons, while the n-propyl cation is a non-stabilised primary ion.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Resonance-energy angle. Hydrolysis of an alkyl chloride
in water is essentially a solvolysis: the slow step is
ionisation to give a carbocation, and the rate scales with the
energy of that cation. The allyl cation, being resonance-delocalised
over two equivalent carbons, gains ∼ 60 kJ/mol of resonance
stabilisation relative to a localised primary cation. By Hammond's
postulate this stabilises the transition state as well, so the
hydrolysis rate of allyl chloride exceeds that of n-propyl
chloride by several orders of magnitude.
Concept used. Solvolysis kinetics of R-Cl in
water follow the SN1 rate law r = k[R-Cl] when
the cation is stable enough to form. For unhindered primary
substrates with no π-neighbour, SN2 takes over;
the comparison here is therefore between an SN1-favoured
allyl chloride and an SN2-favoured n-propyl chloride.
Despite this mechanistic switch, the allyl substrate still wins
on overall hydrolysis rate because its SN1 pathway
is much faster than the n-propyl SN2.
Ionise allyl chloride.CH2=CH-CH2-Cl ->
CH2=CH-CH2+ <-> +CH2-CH=CH2. Charge is shared
across two carbons; transition state is low-energy.
Try ionising n-propyl chloride.CH3CH2CH2-Cl -> CH3CH2CH2+. The primary cation
is unstable and rarely forms; instead the substrate
prefers SN2, which is itself slow because
H2O is a weak nucleophile.
Compare rates. At room temperature in water,
allyl chloride hydrolyses in minutes; n-propyl chloride
is essentially inert under the same conditions.
Resonance-stabilised allyl cation drives faster hydrolysis of allyl chloride by several orders of magnitude over n-propyl chloride.
Q 6.49
Why is it necessary to avoid even traces of moisture during the use of a Grignard reagent?
Concept used. A Grignard reagentR-MgX
has a strongly polarised Cδ--Mgδ+ bond; the
carbon end behaves as a carbanion (very strong base/nucleophile).
Water has a slightly acidic O-H proton (pKa ≈ 15.7),
and any carbanion with conjugate-acid pKa > 16 will deprotonate
water instantly.
Reaction with water: R-MgX + H2O -> R-H + Mg(OH)X.
The Grignard is destroyed; an alkane is formed instead of the
intended addition product.
Even trace moisture (in solvent, glassware, or air) reacts
stoichiometrically, so the yield of the desired product drops
rapidly.
Hence Grignard work must be done in scrupulously dry
ether/THF, under inert (N2 or Ar) atmosphere, with
oven-dried glassware.
R-MgX protolyses with water (R-MgX + H2O -> R-H + Mg(OH)X), destroying the reagent and blocking the desired carbon-carbon bond formation.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Carbanion-protonation angle. The simplest mental model
of a Grignard reagent is ``R- in disguise''. The
C-Mg bond is so polarised that the carbon end behaves as
a strong carbanion-like base, with effective conjugate-acid
pKa near 50 (for R-H). Anything carrying an acidic
proton –- H-O (pKa ≈ 15.7 in water), H-N
(pKa ≈ 36), or even terminal H-C≡C
(pKa ≈ 25) –- is acidic enough to protonate
R-MgX instantly and irreversibly.
Concept used. Acid–base reactivity in organic chemistry
is governed by pKa matching: a base of conjugate-acid pKa
higher than the proton donor's pKa will deprotonate it
quantitatively. R-MgX, with effective pKa near 50,
sits very high on that scale, so virtually any protic species
in the reaction mixture (water, alcohols, amines, terminal
alkynes, carboxylic acids) destroys it. Therefore Grignard work
must use scrupulously dry, aprotic solvents (diethyl ether, THF)
and dry inert atmospheres.
Write the protolysis.R-MgX + H2O -> R-H +
Mg(OH)X. The Grignard donates its carbanion lone pair
to a water proton; the products are an inert alkane and
a Mg(II) hydroxohalide.
Quantify the consequence. Each mole of moisture
destroys one mole of reagent stoichiometrically. Even a
few hundred parts-per-million of H2O in the solvent
is enough to wipe out a synthesis-scale batch.
Hence the precautions. Use dry ether/THF
distilled from Na/benzophenone; oven-dried
glassware; N2 or argon atmosphere; CaCl2
guard tube on the condenser.
R-MgX protolyses with water (R-MgX + H2O -> R-H + Mg(OH)X), destroying the reagent and producing an unreactive alkane in place of the desired addition product.
Q 6.50
Write a test to detect the presence of a double bond in a molecule.
Concept used. A C=C double bond is electron-rich
(π-cloud). Two classic decolourisation tests detect it:
Bromine water test: add a few drops of reddish-brown
Br2 in CCl4 (or aqueous Br2). The alkene
adds Br2 across the π-bond:
R2C=CR2 + Br2 ⟶ R2C(Br)-C(Br)R2.
The reddish-brown colour disappears within seconds. An alkane
does not react and the colour persists.
Baeyer's test (alkaline KMnO4): add dilute
cold alkaline KMnO4 (purple). The alkene is
cis-hydroxylated:
R2C=CR2 + cold dil. KMnO4⟶R2C(OH)-C(OH)R2
The purple colour fades to brown MnO2. Alkanes do not
react under cold conditions.
Either decolourisation ⇒C=C present.
Bromine water (red colour fades) or Baeyer's reagent (purple fades to brown MnO2) detects C=C.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Colour-change angle. The fastest qualitative tests for
a double bond exploit the fact that C=C is electron-rich
(π-cloud) and so reacts cleanly with two coloured reagents
that an alkane would simply ignore. Either reagent reacts via
addition or oxidation at the π-bond, and the visible
endpoint is a sudden colour change that you can describe in a
single sentence on the exam.
Concept used. Two classical wet-lab tests:
Bromine water test (electrophilic addition).
Br2/CCl4 is reddish-brown. The alkene adds
Br2 across the double bond via a cyclic bromonium
intermediate, giving a colourless vicinal dibromide;
the reddish-brown disappears in seconds.
Baeyer's test (cis-hydroxylation). Cold,
dilute, alkaline KMnO4 is deep purple. The
alkene is oxidised to a syn-diol; manganese is reduced
to MnO2, a brown precipitate. The purple fades
and a brown solid appears.
Set up bromine water test. A few drops of
reddish-brown Br2 in CCl4 are added to the
unknown sample.
R2C=CR2 + Br2 ⟶ R2C(Br)-C(Br)R2.
Colour vanishes ⇒ alkene confirmed.
Run Baeyer's test as confirmation. Add cold
dilute KMnO4/NaOH:
R2C=CR2 + cold dil. KMnO4⟶R2C(OH)-C(OH)R2
Purple fades to brown MnO2 precipitate.
Interpret results. Either decolourisation
(better: both) is positive for C=C. An alkane
leaves both reagents unchanged.
Decolourisation of reddish-brown Br2/CCl4 or fading of purple alkaline KMnO4 (with brown MnO2 appearing) confirms the presence of C=C.
Q 6.51
Why are aryl halides less reactive towards nucleophilic substitution than alkyl halides?
Concept used. Three structural features deactivate aryl
halides toward nucleophilic substitution.
Resonance shortening of C-X. A lone pair
on X delocalises into the ring, giving the C-X
bond partial double-bond character. Shorter, stronger
bonds break less easily. Bond lengths: C-Cl in
chlorobenzene ≈ 169 pm vs ≈ 178 pm in
CH3Cl.
Hybridisation of the ipso carbon. The aryl carbon
is sp2 (more s-character, 33%); sp2–halogen
bonds are stronger than sp3–halogen bonds because s
orbitals are closer to the nucleus.
Repulsion in the transition state. The incoming
nucleophile and the electron-rich π-cloud of the ring
repel each other, raising Ea for both SN1
(no stable aryl cation) and SN2 (back-side
attack blocked).
To enhance reactivity, introduce strong electron-withdrawing
groups (-NO2, -CN, -COOH) at o- and p-positions.
The NO2 group stabilises the Meisenheimer intermediate by
resonance, opening up the addition–elimination (SNAr)
pathway. E.g. 2,4-dinitrochlorobenzene reacts with aqueous NaOH
at ∼ 100∘C to give the corresponding phenol;
chlorobenzene itself needs 300∘C + 300 atm (Dow
process).
Aryl halides are unreactive due to resonance shortening of C-X, sp2 carbon hybridisation, and nucleophile–π repulsion. Reactivity is boosted by adding -NO2 groups at o/p-positions.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Three-pillar angle. The unreactivity of aryl halides
rests on three independent structural facts which we can label
the ``three pillars'': (1) resonance shortens and strengthens
the C-X bond, (2) the ipso carbon is sp2 (higher
s-character) so its bond to X is intrinsically stronger
than sp3, and (3) the transition states for both
SN1 and SN2 are destabilised –- the aryl
cation is high in energy and back-side attack on the ipso carbon
is geometrically blocked by the ring's π-cloud. Activating
the substrate with NO2 groups at o and p positions
opens a fourth pathway, the addition–elimination
SNAr, which routes around all three obstacles.
Concept used. The SNAr mechanism for an
activated aryl halide proceeds via a discrete
Meisenheimer intermediate: the nucleophile adds to the
ipso carbon to give an sp3-hybridised carbanion delocalised
over the ring, then loss of X- regenerates aromaticity.
Strong -M groups (-NO2, -CN, -COR) at o/p
positions stabilise the negative charge in the intermediate by
resonance; m- substituents cannot reach the carbanion lobes
and so provide no acceleration. The textbook benchmarks are
300∘C/300 atm for unactivated chlorobenzene (Dow
process) versus 100∘C/1 atm for 2,4-dinitrochlorobenzene.
Pillar 1 – Resonance shortening. In chlorobenzene
the Cl lone pair conjugates into the ring, giving
C-Cl partial double-bond character. Bond lengths:
C-Cl (PhCl) ≈ 169 pm vs ≈ 178 pm
in CH3Cl –- the shorter, stronger bond resists
cleavage.
Pillar 2 – Hybridisation. The aryl carbon is
sp2 (33% s); sp2–halogen bonds are stronger
and shorter than sp3–halogen bonds because s
electrons sit closer to the nucleus.
Pillar 3 – Bad transition states. An aryl
cation would have its empty orbital in the ring plane,
orthogonal to the π-system, so no resonance
stabilisation is possible. SN2 back-side
attack is geometrically forbidden: the ring's π-cloud
sits directly behind the ipso carbon.
Activation via SNAr. Add strong
-M groups at o and p positions (-NO2,
-CN, -COR). The Meisenheimer carbanion is
stabilised by resonance onto those groups; the addition
step is now feasible.
Aryl halides resist classical SN1/SN2 through resonance, hybridisation, and TS effects; activation by o/p-NO2 groups opens the SNAr pathway through a stabilised Meisenheimer intermediate.
Q 6.52
Which of the compounds will react faster in SN1 reaction with the OH- ion: CH3-CH2-Cl or C6H5-CH2-Cl?
Answer.C6H5-CH2-Cl (benzyl chloride) reacts much
faster.
Concept used. The SN1 rate is set by the
ionisation step R-Cl -> R+ + Cl-, so a more stable
R+ means a faster reaction. The benzyl cation
C6H5-C+H2 is resonance-stabilised by four contributors:
C6H5-C+H2 <-> [three ortho/para ring resonance forms]
while the ethyl cation CH3-C+H2 is primary, with no
resonance stabilisation. The benzyl cation is therefore far lower in
energy and forms much faster.
Identify the carbocations: primary ethyl vs benzyl.
Compare stabilisation: ethyl (none) vs benzyl (4 resonance forms).
Conclude: ionisation barrier for benzyl is much lower; SN1 rate is much higher.
Cation-stability angle. The benzyl cation's positive charge
is delocalised onto the o, o', p carbons of the ring, dropping its
energy by ∼ 80 kJ/mol relative to a primary aliphatic cation.
That stabilisation transfers to the TS for ionisation, slashing the
SN1 activation energy.
Concept used.SN1 kinetics: rate = k[R-Cl].
k = A e-Ea/RT, and Ea tracks the energy of the developing
cation. Benzyl < tert < sec < primary < methyl in cation
energy; benzyl is therefore on par with 3∘ for ionisation.
Sketch resonance forms of C6H5CH2+.
Estimate Δ Ea saving ∼ 80 kJ/mol.
Conclude benzyl is the runaway winner.
Benzyl chloride; resonance-stabilised cation.
Q 6.53
Why does iodoform have an appreciable antiseptic property?
Answer. Iodoform (CHI3) is antiseptic because, on
contact with tissues, it slowly liberates free iodine I2.
Iodine is the active germicide; it denatures bacterial proteins by
iodinating tyrosine residues and disrupting essential enzymes.
Concept used. The C-I bond in CHI3 is weak
(∼ 240 kJ/mol) and can be cleaved by skin moisture, enzymes,
or sunlight to liberate I2. The slow release at the wound
gives a sustained antiseptic action.
Apply iodoform paste to the wound.
Body warmth and moisture cleave the weak C-I bonds.
Released I2 kills surface bacteria.
Iodoform's antiseptic action is due to slow liberation of free I2.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Mechanism-of-action angle. The molecule CHI3 itself
has only mild biological activity; the real antiseptic agent is the
slow stream of I2 it releases on the skin or mucous membrane.
Concept used. The C-X bond energy decreases sharply down
the halogen group: C-F ≫ C-Cl > C-Br > C-I.
Iodine is therefore the most easily liberated halide, and iodoform
is essentially a slow-release iodine vehicle.
Confirm C-I in iodoform is the weakest of the C-halide bonds.
Antiseptic due to I2 liberation from the weak C-I bond.
Q 6.54
Discuss the role of Lewis acids in the preparation of aryl bromides and chlorides in the dark.
Answer. Lewis acids such as FeCl3, FeBr3,
AlCl3, AlBr3 generate the active electrophile
X+ by polarising and ultimately abstracting a halide from
X-X:
X2 + FeX3 -> X+ + [FeX4]-
The X+ then attacks the aromatic ring in classical
electrophilic aromatic substitution; the arenium ion is then
deprotonated by [FeX4]-, restoring aromaticity and
regenerating FeX3.
Activation: X2 + FeX3 → X+ ⋯ [FeX4]-.
Electrophilic attack on the ring by X+→ Wheland intermediate (arenium ion).
Loss of H+ to [FeX4]-→ haloarene +HX + FeX3 (catalyst regenerated).
Lewis acids generate X+, the true electrophile in EAS halogenation of arenes.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Catalysis-first angle. Without the Lewis acid, X2
is only weakly polarised by the aromatic π-cloud; activation
energy for direct attack is too high for room-temperature reaction.
The Lewis acid lowers this barrier by pre-polarising X-X and
making one halide effectively cationic.
Concept used. EAS demands an electrophile of sufficient
electrophilicity to overcome the ∼ 150 kJ/mol loss of aromatic
resonance energy in the arenium intermediate. The Lewis acid
catalyst is what makes the electrophile strong enough.
Describe the catalyst's role: X+ generation.
Describe the catalytic cycle: FeX3 regenerated each turn.
Note the ``in the dark'' constraint: light would trigger free-radical
side-chain halogenation instead of ring substitution.
Lewis acids polarise X-X to release X+, the electrophile for EAS.
Q 6.55
Which of the following compounds (a) and (b) will not react with a mixture of NaBr and H2SO4? Explain why.
(a) CH3CH2CH2OH (b) C6H5-OH (phenol)
Answer. (b) phenol does not react with NaBr/H2SO4
to give bromobenzene.
Concept used. The reaction R-OH + HBr -> R-Br + H2O
requires the C-O bond to be cleaved (after protonation of OH).
In phenol the C-O bond has partial double-bond character
because the oxygen lone pair conjugates into the ring; the
C-O is therefore unusually strong and short, and is not
cleaved under these conditions.
(a) n-propanol: C-O is a normal single bond.
H2SO4 protonates OH; Br- (from
NaBr) displaces H2O via SN2→CH3CH2CH2-Br.
(b) Phenol: oxygen lone pair conjugates with the ring; the
C-O bond order is between 1 and 2; bond strength
∼ 460 kJ/mol (vs ∼ 360 in alcohols); not cleaved.
Phenol does not react; C-O in phenol has partial double-bond character (resonance), so it cannot be broken under these conditions.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Resonance angle. The phenolic C-O bond is reinforced
by donation of the O lone pair into the aromatic ring, giving five
resonance contributors and partial double-bond character. The bond
is too strong to cleave with NaBr/H2SO4.
Concept used. Conjugation into an aromatic ring stiffens
the substituent bond. Phenols, anisoles, and aryl amines all show
this behaviour –- their O- or N-substituent bonds are stronger than
the aliphatic analogues.
Sketch phenol resonance structures.
Estimate bond order: >1.
Conclude bond cannot be cleaved by H+/Br- at ordinary conditions.
Phenol unreactive; its C-O is resonance-strengthened.
Q 6.56
Which of the products will be the major product in the reaction given below? Explain. CH3-CH=CH2 + HI -> CH3-CH2-CH2I (A) + CH3-CHI-CH3 (B)
Answer. (B) CH3-CHI-CH3 (2-iodopropane) is the major
product.
Concept used.Markovnikov's rule: in addition of
HX to an unsymmetrical alkene, the H goes to the
carbon with more Hs (terminal CH2) and X goes
to the more substituted carbon (internal CH). Justification:
protonation of propene at the terminal =CH2 gives the more
stable secondary carbocation CH3-C+H-CH3, whereas
protonation at the internal carbon gives the less stable primary
cation CH3-CH2-C+H2.
Two competing protonation sites give either secondary or
primary cation.
Secondary cation is ∼ 80 kJ/mol more stable than
primary ⇒ that path dominates.
I- traps the secondary cation → (B)
2-iodopropane is the Markovnikov product.
Cation-stability shortcut. Pick the more stable cation; the
nucleophile attaches there. For terminal alkenes, that is always the
internal carbon (secondary), so X ends up on C2.
Concept used. Markovnikov regioselectivity is a consequence
of carbocation stability, which is in turn a consequence of
hyperconjugation and +I effects. With peroxides, HBr
(specifically) flips to anti-Markovnikov via radical mechanism; that
exception does not apply to HI or HCl.
Confirm no peroxide → Markovnikov is in force.
Find the secondary cation pathway.
Pick (B).
(B) major; Markovnikov rule.
Q 6.57
Why is the solubility of haloalkanes in water very low?
Answer. Haloalkanes are only weakly polar
(μ ∼ 1.5–2 D), so the dipole-dipole attraction they could
offer to water molecules is weak. More importantly, dissolving them
in water would require breaking the strong hydrogen-bond network of
water, but haloalkanes have no O-H or N-H to replace
those broken H-bonds. The enthalpy cost of disrupting water without
energetic compensation makes the overall solvation unfavourable.
Dissolution would have to (a) break water-water H-bonds and (b) form new water-haloalkane interactions.
Step (a) costs ∼ 20 kJ/mol per H-bond.
Step (b) only repays ∼ 8 kJ/mol per dipole-dipole interaction.
Net Δ H > 0 and Δ S unfavourable (hydrophobic cage) ⇒ low solubility.
Haloalkanes can't H-bond with water; energetically expensive to break water H-bonds without compensation. Hence very low solubility.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Energetics angle. Dissolution is a balance between
solute-solvent attraction (here, weak dipole-dipole) and disruption
of solvent-solvent attraction (here, H-bonds in water). Haloalkanes
lose the second contest.
Low solubility: no H-bonding, weak dipole-dipole interaction.
Q 6.58
Draw resonance structures for halobenzene (C6H5-X, halogen lone pair conjugating with ring), and find out whether the halogen substituent is ortho/para directing or meta directing.
Answer. Halobenzene's -X is ortho/para
directing despite being weakly deactivating overall.
Concept used. A halogen has lone pairs in p-orbitals
which conjugate with the ring's π-system. Resonance structures
place a negative charge (from halogen donation) on the o and p
ring carbons (never on m). This raises electron density at o, p⇒ the electrophile attacks there.
Kekule + 3 charge-separated resonance structures of C6H5-X:
X+ = C ring- on o, p carbons.
Electrophile E+ adds to the C--bearing o, p positions.
No structure puts negative charge on the m carbon
⇒ no m attack.
Halogen is o, p directing because resonance increases the electron density at the ortho and para positions.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Resonance-mapping angle. The lone pair on the halogen
delocalises into the ring through the same π-network used by
benzene's π-electrons. The negative charge it pushes onto the
ring lands on the o and p carbons by symmetry; m is missed
because of the alternating polarity of the ring's π-system.
Concept used. Two opposing effects: halogen -I withdraws
σ-electrons (deactivating), halogen +M pushes lone pairs
(activating). The -I effect wins on average rate; the +M effect
selects o, p positions.
Draw the three charge-separated resonance forms putting
C- at o, o', p.
Conclude o, p-directing.
Note: deactivating despite directing o, p.
o, p-directing through +M resonance; weakly deactivating through -I.
Q 6.59
Classify the following compounds as primary, secondary and tertiary halides:
(i) 1-Bromobut-2-ene (ii) 4-Bromopent-2-ene (iii) 2-Bromo-2-methylpropane
Answers.
(i) Primary; (ii) Secondary; (iii) Tertiary.
Concept used. The class of an alkyl halide is set by the
number of other carbon atoms attached to the C-X
carbon: 1 → primary, 2 → secondary, 3 → tertiary.
Substituent-count angle. For each substrate, draw the
skeletal structure and count carbons bonded to the C-X
carbon.
Concept used. The class only counts carbon neighbours; the
identity of those carbons (alkyl, vinyl, aryl) doesn't change the
class but does affect reactivity (e.g. allyl primary halides
ionise like tertiary halides because of resonance).
Sketch each structure.
Count carbons on C-X.
Assign 1∘, 2∘, 3∘.
(i) 1∘; (ii) 2∘; (iii) 3∘.
Q 6.60
Compound `A' with molecular formula C4H9Br is treated with aq. KOH solution. The rate of this reaction depends upon the concentration of the compound `A' only. When another optically active isomer `B' of this compound was treated with aq. KOH solution, the rate of reaction was found to be dependent on concentration of compound and KOH both.
(i) Write down the structural formula of both compounds `A' and `B'.
(ii) Out of these two compounds, which one will be converted to the product with inverted configuration?
Answers.
(i) A: (CH3)3C-Br (tert-butyl bromide; rate ∝ [A]
only ⇒SN1, so A is tertiary).
B: CH3-CHBr-CH2-CH3 (2-bromobutane; rate ∝
[B][KOH] ⇒SN2, so B is secondary and optically active).
(ii) Compound B (because SN2 proceeds with Walden
inversion).
Concept used. Rate law identifies the mechanism: first-order
⇒SN1 (substrate must be tertiary or
benzyl/allyl), second-order ⇒SN2 (substrate
typically primary or secondary). SN2 inverts; SN1
racemises.
A is the only C4H9Br that gives a tertiary cation =(CH3)3CBr.
B is optically active ⇒ chiral; the only chiral C4H9Br is 2-bromobutane.
B reacts SN2⇒ Walden inversion ⇒ inverted product.
A = (CH3)3CBr; B = CH3CHBrCH2CH3; B gives inverted product.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Kinetics-reads-mechanism angle. Order of reaction is the
single most powerful tool for distinguishing SN1 vs
SN2. First-order ⇒ unimolecular ionisation
step is rate-limiting ⇒ no OH- in rate law.
Second-order ⇒ both substrate and nucleophile are in
the rate-determining step.
Concept used. Stereochemistry follows mechanism:
SN1 gives racemisation (planar carbocation, equal
attack from either face); SN2 gives inversion (back-side
attack).
Read rate law → mechanism.
Match substrate skeleton.
Apply stereo outcome.
A is 3∘ (SN1, racemises); B is 2∘ (SN2, inverts).
Q 6.61
Write the structures and names of the compounds formed when compound `A' with molecular formula C7H8 is treated with Cl2 in the presence of FeCl3.
Answer. A = toluene (C6H5-CH3). EAS chlorination with
Cl2/FeCl3 gives a mixture of o-chlorotoluene and
p-chlorotoluene (with p as major).
Concept used.-CH3 is an o/p-director (activating,
+I/hyperconjugation). FeCl3 generates Cl+ which
attacks the ring; the methyl group steers the electrophile to o
and p.
Generate Cl+: Cl2 + FeCl3 -> Cl+ + [FeCl4]-.
Methyl stabilises positive charge in the σ-complex
when Cl adds at o or p.
Products: o-chlorotoluene (2-chlorotoluene) and
p-chlorotoluene (4-chlorotoluene).
Structures: o-Cl-C6H4-CH3 and p-Cl-C6H4-CH3
o- and p-chlorotoluene.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Director-first angle. ``C7H8'' and ``Lewis acid + Cl''
together name toluene + EAS chlorination. The methyl group
predictably steers the electrophile to ortho and para.
Concept used. Activating + ortho/para-directing groups
(CH3, OH, NH2, OR) all give mixtures of o and p
products with p usually dominant (less steric clash).
Identify A as toluene.
Predict EAS products with methyl as o/p-director.
Draw both isomers.
o- and p-chlorotoluene.
Q 6.62
Identify the products A and B formed in the following reaction: CH3CH2-CH=CH-CH3 + HCl -> A + B
Answer.
A = CH3CH2-CH2-CHCl-CH3 (2-chloropentane).
B = CH3CH2-CHCl-CH2-CH3 (3-chloropentane).
Concept used. The alkene is pent-2-ene
(CH3CH2-CH=CH-CH3). Both sp2 carbons of the double bond
have one H each ⇒ Markovnikov's rule doesn't
discriminate (both possible cations are secondary). Therefore two
products form in comparable amounts.
Protonate the C=C at C2→ secondary cation CH3CH2-CH2-C+H-CH3.
Or protonate at C3→ different secondary cation CH3CH2-C+H-CH2-CH3.
Both are secondary; both form. Cl- traps each → A and B.
A = 2-chloropentane; B = 3-chloropentane.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Markovnikov-degeneracy angle. When both alkene carbons give
the same class of cation, Markovnikov's rule loses its
predictive power. Both products are expected.
Concept used. Internal symmetric or near-symmetric alkenes
yield regiochemical mixtures because no carbocation is significantly
more stable than the other.
Sketch both cation pathways.
Both secondary, near-isoenergetic.
Both products form.
Mixture of 2-chloro- and 3-chloropentane.
Q 6.63
Which of the following compounds will have the highest melting point and why?
(I) 1,3-Dichloro-2,5-dimethylbenzene
(II) 2,5-Dichloro-1,4-dimethylbenzene
(III) 1,3-Dichloro-2,4-dimethylbenzene
Answer. (II) 2,5-Dichloro-1,4-dimethylbenzene has the
highest melting point.
Concept used. Melting point of substituted benzenes
correlates strongly with the symmetry of the molecule:
more symmetric isomers pack tightly into the crystal lattice and
require more energy to disrupt. (II) has C2h symmetry (1,4-
methyls + 2,5- chlorines all related by the centre of inversion);
(I) and (III) lack this high symmetry.
Inspect the three substitution patterns.
(II) has both pairs of substituents at p-positions to each
other ⇒ highest symmetry ⇒ tightest packing.
(II) has the highest m.p.
(II) has the highest m.p. because of its p-symmetric, tightly-packing structure.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Crystal-packing angle. The 1,4 + 2,5 substitution pattern
makes (II) the only one whose unit cell can be a near-cubic dense
packing without steric clash; the others have at least one ortho
clash that loosens the lattice.
Concept used. Lattice energy ∝ symmetry and packing
density. More symmetry ⇒ better packing ⇒
higher m.p.
Assess symmetry of each isomer.
Highest-symmetry isomer wins.
(II) highest m.p. owing to highest molecular symmetry.
Q 6.64
Write down the structure and IUPAC name for neo-pentyl bromide.
Answer. Structure: (CH3)3C-CH2Br (3,3-dimethyl-1-bromopropane? No –- the longest chain is propane with two methyls on C2). IUPAC name: 1-Bromo-2,2-dimethylpropane.
Concept used. ``Neopentyl'' = (CH3)3C-CH2- (a
C5 fragment with three methyls on the carbon adjacent to the
CH2 being substituted). IUPAC: parent is propane (the longest
chain through the C-Br carbon), substituents are two methyls
at C2 and Br at C1.
Draw neopentyl group: (CH3)3C-CH2-.
Attach Br: (CH3)3C-CH2Br.
Find longest chain: 3 C (propane) through CH2-C-CH3.
Trivial-to-IUPAC translation. ``Neo'' (Greek for ``new'')
labels a five-carbon fragment with three methyls on one terminal C.
For IUPAC, find the longest chain through the substituted carbon –-
it is 3 carbons (the CH2, then the quaternary C, then one of
the three methyls), not 4.
Cite ``bromo'' before ``dimethyl'' alphabetically.
1-Bromo-2,2-dimethylpropane.
Q 6.65
A hydrocarbon of molecular mass 72 g mol-1 gives a single monochloro derivative and two dichloro derivatives on photochlorination. Give the structure of the hydrocarbon.
Answer. The hydrocarbon is neo-pentane,
(CH3)4C (2,2-dimethylpropane).
Concept used. Molecular mass 72 ⇒C5H12
(5× 12 + 12 = 72). Among the three isomers of pentane
(n-pentane, isopentane, neopentane), only neopentane has
all 12 hydrogens in equivalent positions; therefore only one
monochloro derivative forms. The second chlorination, on the
monochloro derivative (CH3)3C-CH2Cl, has two distinct
positions: (a) the same CH2Cl carbon (gem) and (b) one of the
three remaining methyl groups, giving exactly two dichloro
derivatives.
Mass 72 →C5H12 pentane isomer.
Single monochloro → all H's equivalent → neopentane.
Monochloro derivative: (CH3)3C-CH2Cl.
Two further dichloro positions: gem (same carbon) or distal methyl.
Hydrocarbon: (CH3)4C (neopentane).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Symmetry-counts-isomers angle. The number of distinct
monochloro products equals the number of inequivalent H-bearing
positions in the parent. Neopentane has just one such position; it
must be the answer.
Concept used. Hydrogen equivalence under molecular
symmetry; molecular formula from molecular mass.
Compute formula from mass.
List pentane isomers.
Pick the one with all H's equivalent.
Neopentane, (CH3)4C.
Q 6.66
Name the alkene which will yield 1-chloro-1-methylcyclohexane by its reaction with HCl. Write the reactions involved.
Answer. Either methylenecyclohexane
(C6H10=CH2, exocyclic alkene) or
1-methylcyclohex-1-ene (endocyclic alkene with CH3 on C1)
yields 1-chloro-1-methylcyclohexane.
Concept used. Markovnikov addition of HCl goes
through the most stable carbocation. In both cases the most stable
cation is the tertiary cation centred on the ring carbon that bears
the methyl group. Cl- then traps that tertiary carbon
⇒ both starting alkenes converge on the same product.
Methylenecyclohexane + HCl:
H+ adds to the =CH2, generating the
tertiary cation at the ring carbon; Cl- adds there
→ 1-chloro-1-methylcyclohexane.
1-Methylcyclohex-1-ene + HCl:
H+ adds to C2 of the ring, generating the
tertiary cation at C1; Cl- adds there
→ same product.
Both methylenecyclohexane and 1-methylcyclohex-1-ene give 1-chloro-1-methylcyclohexane via Markovnikov addition through a tertiary cation.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Same-cation angle. Two structurally distinct alkenes can
give the same product if they share an intermediate carbocation.
Here both alkenes protonate to the same tertiary cation
(``1-methylcyclohexyl cation''), which then captures Cl-.
Concept used. Markovnikov regioselectivity is governed by
cation stability. Tertiary cations win over secondary or primary,
so the proton goes wherever leaves a tertiary cation behind.
For each alkene, draw the more stable cation.
Note that both cations are the same species.
Cl- traps → same product.
Methylenecyclohexane or 1-methylcyclohex-1-ene.
Q 6.67
Which of the following haloalkanes reacts with aqueous KOH most easily? Explain giving reason.
(i) 1-Bromobutane (ii) 2-Bromobutane
(iii) 2-Bromo-2-methylpropane (iv) 2-Chlorobutane
Answer. (iii) 2-Bromo-2-methylpropane (tert-butyl bromide)
reacts most easily.
Concept used. With aq. KOH (polar protic solvent, good
ionising medium, weak nucleophile OH- at low concentration
relative to water), the dominant mechanism for tertiary halides is
SN1 via the tertiary carbocation. The tert-butyl cation
(CH3)3C+ is highly stabilised by hyperconjugation and +I,
so the ionisation barrier is low. Additionally, Br is a
better leaving group than Cl.
Tertiary substrate ⇒SN1 via stable
tertiary cation.
Br leaves more readily than Cl (weaker C-X bond, more polarisable).
Combined: tertiary bromide is fastest.
(iii) 2-Bromo-2-methylpropane: tertiary + Br leaving group ⇒ fastest.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Two-factor angle. Two variables decide the rate: (1) class
of the halide (cation stability) and (2) leaving group quality.
Tertiary bromide wins on both axes.
Why can aryl halides not be prepared by reaction of phenol with HCl in the presence of ZnCl2?
Answer. The C-O bond in phenol has partial double-bond
character because the oxygen lone pair conjugates with the aromatic
ring. This stabilised, shortened bond is not cleaved by
HCl/ZnCl2, so Cl cannot displace OH from
the ring.
Phenolic C-O enjoys resonance stabilisation
(∼ 460 kJ/mol vs ∼ 360 in aliphatic alcohols).
ZnCl2 would normally protonate OH and weaken
C-O, allowing Cl- to displace water.
In phenol the partial double-bond character resists cleavage
⇒ no reaction.
Phenol's C-O has partial double-bond character (resonance) and cannot be broken by HCl/ZnCl2.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Resonance-strengthens-bond angle. The same resonance that
makes phenol acidic also makes the C-O bond very hard to
break in the wrong direction. Lucas-type reagents fail; even
PCl5 gives at best triphenyl phosphate, not chlorobenzene.
Concept used. Bond order >1 implies bond energy
significantly above single-bond baseline. Cleavage requires
extraordinary conditions (e.g. diazotisation route via
ArN2+).
Cite phenol's resonance forms putting C=O+H
character on the ipso carbon.
Conclude C-O is too strong to break.
Note alternative: C6H5-NH2 -> C6H5-N2+ -> C6H5-Cl
via Sandmeyer.
Resonance makes phenolic C-O unbreakable by Lucas-type reagents.
Q 6.69
How do polar solvents help in the first step in SN1 mechanism?
Answer. Polar solvents (especially polar protic ones like
water, methanol) accelerate the first step of SN1 –-
the ionisation R-X -> R+ + X- –- by solvating
both the developing carbocation and the leaving halide ion. The
solvation lowers the energy of the ion pair, reduces the activation
energy for ionisation, and makes the heterolysis viable at room
temperature.
Polar solvent molecules orient their negative ends around
R+ (cation solvation) and their positive ends (H of
OH) around X- (anion solvation, via H-bonding).
Solvation energy of Cl- in water is ∼ -380 kJ/mol;
similar magnitude for R+.
This stabilisation drops the ionisation barrier from
∼ 250 kJ/mol (gas phase) to ∼ 100 kJ/mol (solution).
Ionisation now occurs at observable rate.
Polar protic solvents solvate R+ and X-, dropping the ionisation energy and enabling SN1.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Solvation-stabilisation angle. The rate-determining step of
SN1 is heterolysis; this step creates ions.
Without a medium to stabilise those ions, the reaction wouldn't
happen at room temperature. Polar protic solvents are tailor-made
for this stabilisation.
Concept used. Dielectric constant ε governs how
much the solvent reduces electrostatic attraction between newly
formed ions; H-bonding capacity governs anion stabilisation;
both contribute to lowering Ea for ionisation.
Identify the ionisation step.
Identify what is needed: stabilisation of the new ions.
Match: polar protic solvent (high ε + H-bonding).
Solvation of R+ and X- lowers Ea for ionisation.
Q 6.70
Diphenyls are potential threat to the environment. How are these produced from aryl halides?
Answer. Diphenyl (biphenyl, C6H5-C6H5) and its
chlorinated derivatives (PCBs, polychlorinated biphenyls) are
produced by the Fittig reaction: an aryl halide is
treated with sodium metal in dry ether:
2 C6H5-X + 2 Na ->[dry ether] C6H5-C6H5 + 2 NaX
The polychlorinated analogues (e.g. from chlorobenzene under
industrial conditions, or from accidental coupling during PCB
manufacture) are highly persistent organic pollutants and
bioaccumulate.
2 Ar-X + 2 Na -> Ar-Ar + 2 NaX (Fittig).
For chlorobenzene → biphenyl + 2 NaCl.
Chlorinated biphenyls (PCBs) are non-biodegradable and lipophilic;
accumulate in food chain (Stockholm convention banned).
Diphenyls form by the Fittig reaction of aryl halides with Na in dry ether; PCBs are the toxic chlorinated variants.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Named-reaction angle. The Fittig reaction is the aryl-aryl
analogue of Wurtz (which couples two alkyl halides). The mechanism
goes through aryl radicals or aryl anions on the sodium surface.
Concept used. Reductive coupling: two C-X bonds break,
one C-C bond forms; the metal (Na) provides electrons.
Identify the coupling: aryl-aryl → biphenyl.
Cite Fittig conditions: Na in dry ether.
Note environmental relevance of PCBs.
Fittig reaction; biphenyls/PCBs.
Q 6.71
What are the IUPAC names of the insecticide DDT and benzene hexachloride? Why is their use banned in India and other countries?
Why banned: both are persistent, fat-soluble organochlorine
pesticides; they bioaccumulate in the food chain, harm
non-target species (birds, fish, beneficial insects), and cause
endocrine disruption and probable carcinogenicity in humans.
BHC structure: cyclohexane with one Cl on each of
six carbons (γ-isomer is the active Lindane).
IUPAC: 1,2,3,4,5,6-hexachlorocyclohexane.
Persistence: low water solubility, high lipid solubility;
half-life in soil ∼ years.
Banned because of bioaccumulation, food-chain magnification
(eggshell thinning in raptors), and chronic toxicity.
DDT = 1,1,1-trichloro-2,2-bis(4-chlorophenyl)ethane; BHC = 1,2,3,4,5,6-hexachlorocyclohexane. Banned due to persistence and bioaccumulation.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Persistent-organic-pollutant angle. DDT and BHC were
celebrated mid-20th-century insecticides; the very C-Cl
bonds that made them effective also made them unbreakable in the
environment.
Concept used. Organochlorine compounds, with several
C-Cl bonds, resist enzymatic degradation. Their lipid
solubility (log P > 5) drives accumulation in fatty tissue;
biomagnification climbs each trophic level.
Elimination reactions (especially β-elimination) are as common as the nucleophilic substitution reaction in case of alkyl halides. Specify the reagents used in both cases.
Elimination: alc. KOH or t-BuOK (alkene by E2);
no nucleophile, just heat (E1 of tert halides).
Concept used. Solvent and base bulk select the partition.
Aqueous + small base ⇒ SN; alcoholic + small
base ⇒ E2; bulky base regardless of solvent ⇒
E2.
List reagent pairs by mechanism.
Note the solvent dependence.
As tabulated above.
Q 6.73
How will you obtain monobromobenzene from aniline?
Answer. Diazotise aniline, then perform a Sandmeyer
reaction with Cu2Br2:
(Or use HBF4 for the Schiemann variant; for bromide, the Cu salt
is the cheap textbook route.)
Concept used. Aniline is too activated for direct
bromination (Br2 alone gives 2,4,6-tribromoaniline); the
diazonium route gives clean mono-substitution.
Diazotise: NaNO2 + HCl at 0–5 ∘C →C6H5N2+ Cl-.
Treat with Cu2Br2 (Sandmeyer) →C6H5-Br + N2 + CuCl.
Pure monobromobenzene is isolated.
Aniline → diazonium → Sandmeyer with Cu2Br2 gives bromobenzene.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Diazonium-as-handle angle. The diazonium salt
ArN2+ is one of the most versatile intermediates in
synthesis. From it you can install -F, -Cl, -Br, -CN, -OH,
-H, -I, -NO2, -SH, -Ar on the ring with the appropriate Cu salt
or hot water etc.
Concept used. Sandmeyer-type radical substitution converts
N2+ to X with loss of N2. Cu(I) salt acts
as a one-electron shuttle.
Diazotise aniline.
Cu2Br2 Sandmeyer to install Br.
Aniline → ArN2+→ PhBr by Sandmeyer.
Q 6.74
Aryl halides are extremely less reactive towards nucleophilic substitution. Predict and explain the order of reactivity of the following compounds towards nucleophilic substitution:
(I) p-nitrochlorobenzene
(II) 2,4-dinitrochlorobenzene
(III) 2,4,6-trinitrochlorobenzene
Answer. Reactivity: III > II > I.
Concept used. Each additional o/p-NO2 group
provides another resonance sink for the negative charge in the
Meisenheimer adduct of the SNAr pathway. More NO2
groups ⇒ more stabilisation ⇒ faster rate.
(I) one p-NO2: moderate activation. Reacts with aq. NaOH at ∼ 100∘C.
(II) two NO2 at 2,4 (one o, one p): much faster.
Reacts with aq. NaOH at ∼ 100∘C in dilute conditions.
(III) three NO2 at 2,4,6 (two o, one p): picryl chloride.
Reacts with H2O alone at room temperature.
Reactivity order: III > II > I.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Multi-NO2 stacking angle. Three nitros suitably placed
turn an inert aryl halide into one that reacts with water alone at
room temperature –- a rate boost of ∼ 1012.
Concept used. Successive electron-withdrawing groups
operate near-independently in stabilising the Meisenheimer
intermediate; rate scales as roughly the product of single
activation factors.
Count o, p nitros in each substrate.
Order by count: III (3) > II (2) > I (1).
III > II > I.
Q 6.75
tert-Butyl bromide reacts with aq. NaOH by SN1 mechanism while n-butyl bromide reacts by SN2 mechanism. Why?
Answer. The two halides differ in the class of the
substrate carbon and therefore in (a) carbocation stability and (b)
steric access to the substrate carbon.
tert-Butyl bromide (CH3)3CBr: tertiary
substrate. (i) The SN1 cation
(CH3)3C+ is highly stabilised by three +I donors
and nine α-H hyperconjugations. (ii) The substrate
carbon is sterically blocked by three methyl groups,
preventing back-side SN2 attack. Both factors
force SN1.
n-Butyl bromide CH3CH2CH2CH2Br: primary
substrate. (i) The SN1 cation
CH3CH2CH2-C+H2 is primary and very unstable
(no +I/hyperconjugation buffer worth the name). (ii)
Back-side SN2 attack on CH2Br is sterically
unobstructed. Both factors force SN2.
Tertiary substrate stabilises carbocation and blocks back-side attack ⇒SN1; primary substrate is opposite on both axes ⇒SN2.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Two-axis angle. Mechanism is dictated by the two opposing
demands: cation stability (favours SN1 for tertiary) and
back-side accessibility (favours SN2 for primary). Each
substrate falls on the opposite extreme.
Concept used. Carbocation stability scale
3∘ > 2∘ > 1∘ > CH3+; steric crowding
inverts the order for SN2 accessibility.
Tag substrate class.
Look up cation stability and steric access.
Assign mechanism.
tert-Bu-Br → SN1; n-Bu-Br → SN2.
Q 6.76
Predict the major product formed when HCl is added to isobutylene. Explain the mechanism involved.
Answer. Major product: 2-chloro-2-methylpropane
((CH3)3C-Cl).
Concept used. Markovnikov addition of HCl to the
unsymmetrical alkene (CH3)2C=CH2 (isobutylene) proceeds via
the more stable carbocation.
Mechanism.
Step I:H+ protonates the =CH2
terminus → tertiary cation (CH3)3C+ (very
stable). The alternative would be the primary cation
(CH3)2CH-C+H2 –- discarded because it's much
higher energy.
Step II:Cl- traps the tertiary cation
→(CH3)3C-Cl (2-chloro-2-methylpropane).
Markovnikov product (CH3)3C-Cl; via tertiary cation intermediate.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Cation-stability angle. Out of the two possible cations
(3∘ vs 1∘), the tertiary is ∼ 100 kJ/mol more
stable; the regioselectivity is essentially complete.
Concept used. Markovnikov regioselectivity is set by
carbocation stability; for terminal disubstituted alkenes, the
proton always adds to give the more substituted cation.
Identify the two possible cations.
Pick the tertiary.
Cl- traps it.
(CH3)3C-Cl, Markovnikov.
Q 6.77
Discuss the nature of C-X bond in the haloarenes.
Answer. The C-X bond in haloarenes is shorter and
stronger than the corresponding bond in haloalkanes because of two
effects:
1pt
Hybridisation: the ring carbon is sp2 (33% s),
while an alkyl C is sp3 (25% s). Higher s-character
places the bonding electrons closer to the nucleus and
shortens the bond.
Resonance: the halogen lone pair conjugates with
the ring's π-system, putting partial double-bond
character on C-X. Bond order >1⇒
shorter, stronger.
Compare bond lengths: chlorobenzene C-Cl∼ 169 pm vs methyl chloride ∼ 178 pm.
Compare bond energies: PhCl ∼ 400 kJ/mol vs MeCl ∼ 339 kJ/mol.
Conclude: stronger bond ⇒ less reactive in nucleophilic substitution.
Aryl C-X is shorter and stronger than alkyl C-X: sp2 + resonance.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Hybridisation-plus-resonance angle. Two cooperative effects
reinforce each other: more s-character in the bonding orbital
(geometry) AND donation from the halogen lone pair into the ring
(π-character). Both shorten and strengthen the bond.
Concept used. Bond length and bond strength are inversely
correlated. Hybridisation s-character: sp > sp2 > sp3 in
bond strength. Resonance donation: introduces partial double-bond
character.
Cite hybridisation.
Cite resonance.
Quantify with bond-length and bond-energy figures.
Aryl C-X is shorter and stronger; partial double-bond character + sp2.
Q 6.78
How can you obtain iodoethane from ethanol when no other iodine-containing reagent except NaI is available in the laboratory?
Answer. Two-step sequence: first convert ethanol to a
suitable haloalkane (the chloride), then run Finkelstein with NaI.
C2H5-OH + HCl/ZnCl2 -> C2H5-Cl + H2O
C2H5-Cl + NaI ->[dry acetone] C2H5-I + NaCl↓
Concept used. Direct conversion of R-OH to R-I
requires HI (not available). Instead, make the more accessible
chloride first using HCl/ZnCl2 (Lucas reagent), then
exchange Cl for I using the Finkelstein
reaction: in dry acetone, NaCl precipitates (insoluble in
acetone) and shifts the equilibrium toward R-I.
Lucas: C2H5-OH + HCl/ZnCl2 -> C2H5-Cl + H2O.
Finkelstein: C2H5-Cl + NaI ->[dry acetone] C2H5-I + NaCl.
NaCl precipitates from acetone, driving equilibrium right.
Ethanol → ethyl chloride (Lucas) → ethyl iodide (Finkelstein with NaI in dry acetone).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Two-step retrosynthesis. Disconnect C2H5-I to
C2H5-Cl via halide exchange; disconnect C2H5-Cl to
C2H5-OH via Lucas. The trick is recognising that the only
I source is NaI, and Finkelstein is the only practical way
to use it.
Concept used. Finkelstein equilibrium is driven to
completion by solubility differences: NaCl insoluble in
acetone, NaI soluble.
Convert ethanol to ethyl chloride (Lucas).
Convert ethyl chloride to ethyl iodide (Finkelstein).
Two-step: Lucas then Finkelstein.
Q 6.79
Cyanide ion acts as an ambident nucleophile. From which end does it act as a stronger nucleophile in aqueous medium? Give reason for your answer.
Answer. In aqueous medium, CN- attacks
through carbon as the stronger nucleophile, giving alkyl
cyanides (R-C#N, nitriles), not alkyl isocyanides.
Concept used.CN- has lone pairs on both C and N
(ambident). The C end is softer/more polarisable and forms a
stronger covalent C-C bond (∼ 350 kJ/mol) than the
C-N bond (∼ 290 kJ/mol) that would result from N-attack.
In water, where the harder N end is more solvated by
H-bonding, the C end is even more exposed and reactive.
Identify ambident character: C and N both nucleophilic.
C-end is the stronger nucleophile; C-C bond is more stable than C-N, and C is less solvated in water.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Ambident-thermodynamics angle. The cyanide anion has two
nucleophilic atoms with different hardness. In an ionising medium
(water), the softer C end dominates because of the stronger C-C
bond formed and weaker solvation of the C lone pair.
Concept used. Hard-Soft Acid-Base (HSAB) theory: hard
nucleophiles prefer hard electrophiles; soft prefer soft. Alkyl
halides at sp3 C are intermediate, leaning soft; C of cyanide
matches.
Identify both ends as nucleophilic.
Compare bond-strength of products: C-C > C-N.
Compare solvation in water: N more solvated than C.
Conclude C-attack wins.
C end stronger nucleophile in aqueous medium.
IV. Matching Type
Q 6.80
Match the compounds given in Column I with the effects given in Column II. [2pt]
tabularp0.42p0.45
Column I & Column II
(i) Chloramphenicol & (a) Malaria
(ii) Thyroxine & (b) Anaesthetic
(iii) Chloroquine & (c) Typhoid fever
(iv) Chloroform & (d) Goiter
& (e) Blood substituent
tabular
Matches. (i)→(c); (ii)→(d); (iii)→(a); (iv)→(b).
Concept used. Each halogen-containing compound has a
characteristic physiological action used in medicine.
(i) Chloramphenicol is an antibiotic given for
typhoid fever caused by Salmonella typhi.
(ii) Thyroxine (T4), an iodine-rich hormone secreted
by the thyroid; its deficiency leads to enlargement of
thyroid gland ⇒goiter.
(iii) Chloroquine is the classical drug for
malaria (treats P. vivax, P. falciparum).
(iv) Chloroform (CHCl3) was historically used as an
anaesthetic (now replaced by safer halothane/isoflurane).
(i)→(c); (ii)→(d); (iii)→(a); (iv)→(b).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Drug-class lookup angle. Each name in Column I belongs to
a different therapeutic class. Recall the active indication of each
and match directly.
Concept used. Many haloorganic compounds find use in
medicine because of their bioavailability and specific receptor
interactions: halothane (anaesthetic), chloroquine (antimalarial),
DDT (insecticide –- now banned), iodoform (antiseptic), thyroxine
(hormone), chloramphenicol (antibiotic).
Tag each Column-I compound to its therapeutic class.
Match to the disease in Column II.
As above.
Q 6.81
Match the items of Column I and Column II. [2pt]
tabularp0.42p0.45
Column I & Column II
(i) SN1 reaction & (a) vic-dibromides
(ii) Chemicals in fire extinguisher & (b) gem-dihalides
(iii) Bromination of alkenes & (c) Racemisation
(iv) Alkylidene halides & (d) Saytzeff rule
(v) Elimination of HX from alkyl halide & (e) Chlorobromocarbons
tabular
Concept used. Match each chemical phenomenon to its
diagnostic descriptor.
(i) SN1: proceeds via a planar carbocation;
nucleophile attacks from either face equally, yielding a
50:50 mixture of enantiomers ⇒ (c)
racemisation.
(ii) Fire extinguishers: traditional fillings are
chlorobromocarbons such as CF2ClBr (BCF) and
CF3Br (BTM), prized for free-radical inhibition
⇒ (e).
(iii) Br2 adds across C=C at C1 and
C2 (anti addition through a bromonium ion),
producing Br-CR2-CR2-Br, a vic-dibromide
⇒ (a).
(iv) Alkylidene halides have both halogens on the same
carbon (e.g. CH3-CHCl2, ethylidene chloride)
⇒ (b) gem-dihalides.
(v) Base-induced β-elimination of HX from
R-CHX-CH2R′ obeys the Saytzeff rule: the more
substituted alkene predominates ⇒ (d).
(i)→(c); (ii)→(e); (iii)→(a); (iv)→(b); (v)→(d).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Tag-pairing angle. Each Column I phrase is a one-word
diagnostic descriptor of a phenomenon; Column II offers a
matching keyword. Read both lists, identify the unique pairing
for each row, and you're done. Don't be tempted by superficially
similar pairs (vic/gem look interchangeable but
each belongs to a specific reaction type).
Concept used. Every entry in Column I has a single,
unambiguous chemical signature that appears in Column II.
SN1 via a planar cation → racemisation;
chlorobromocarbons (Halon-type) → fire extinguishers;
Br2 adds anti across C=C→vic-dibromides
through a bromonium intermediate; alkylidene halides are by
definition gem-dihalides (CHX2 shape); base-induced
β-elimination obeys Saytzeff (more substituted alkene
predominates).
(ii) Fire extinguishers. Classical halon
fillings CF2ClBr (Halon 1211, BCF) and
CF3Br (Halon 1301, BTM) suppress flames by
scavenging H. and OH. radicals
⇒(e) Chlorobromocarbons.
(iii) Bromination of alkenes. Anti-addition
through a bromonium ion places Br on C1
and C2⇒(a) vic-dibromides.
(iv) Alkylidene halides. ``-idene'' nomenclature
means both halogens are on a single CH carbon
(CH3-CHCl2, ethylidene chloride) ⇒(b) gem-dihalides.
(v) Elimination of HX. Base abstracts a
β-H to generate the more substituted alkene
(Zaitsev product) ⇒(d) Saytzeff rule.
(i)→(c); (ii)→(e); (iii)→(a); (iv)→(b); (v)→(d).
Q 6.82
Match the structures of compounds given in Column I with the classes of compounds given in Column II. [2pt]
tabularp0.42p0.45
Column I (Structure) & Column II (Class)
(i) CH3-CHX-CH3 & (a) Aryl halide
(ii) CH2=CH-CH2-X & (b) Alkyl halide
(iii) C6H5-X (X on ring carbon) & (c) Vinyl halide
(iv) CH2=CH-X & (d) Allyl halide
tabular
Matches. (i)→(b); (ii)→(d); (iii)→(a); (iv)→(c).
Concept used. Classify halides by the hybridisation /
position of the carbon attached to X:
(ii) CH2=CH-CH2-X: sp3 C next to C=C→allyl halide (d).
(iii) C6H5-X: X on aromatic ring →aryl halide (a).
(iv) CH2=CH-X: sp2 C of C=C→vinyl halide (c).
(i)→(b); (ii)→(d); (iii)→(a); (iv)→(c).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Position-of-X angle. The four canonical halide classes
differ only in where the halogen sits relative to
unsaturation: alkyl (away from any π), allyl (next to C=C),
vinyl (on C=C), aryl (on aromatic ring). Reactivity scales
predictably.
Concept used. Reactivity tags:
1pt
Allyl/benzyl ⇒ fast SN1 (resonance-stabilised cation).
Alkyl ⇒ standard SN1/SN2 depending on class.
Vinyl/aryl ⇒ unreactive in classical SN
(partial double bond + sp2 carbon).
Identify hybridisation/location of the C-X carbon.
Pick the corresponding class.
As tabulated.
Q 6.83
Match the reactions given in Column I with the types of reactions given in Column II. [2pt]
tabularp0.52p0.4
Column I (Reaction) & Column II (Type)
(i) Chlorobenzene + Fe/Cl2→o- and p-dichlorobenzene & (a) Nucleophilic aromatic substitution
(ii) Propene + HBr → 2-bromopropane & (b) Electrophilic aromatic substitution
(iii) C6H5-CHI-CH3 + OH-→C6H5-CHOH-CH3 & (c) Saytzeff elimination
(iv) p-nitrochlorobenzene + NaOH →p-nitrophenol & (d) Electrophilic addition
(v) 2-Bromobutane + alc. KOH → 2-butene & (e) Nucleophilic substitution (SN1)
tabular
Concept used. Tag each reaction by what attacks what:
electrophile attacks aromatic ring → EAS; nucleophile displaces
LG on aromatic ring with -M activation → SNAr; H+ adds
across C=C→ electrophilic addition; benzyl halide hydrolysed
in water → SN1 via stable cation; base abstracts β-H
to give more substituted alkene → Saytzeff E2.
(i) Fe/Cl2 on PhCl: ring substitution by Cl+→ EAS (b).
(ii) HBr to propene: H+ adds, then Br- at secondary carbon →
electrophilic addition (d).
Reaction-type tagging angle. For every transformation, ask
``what is the substrate (aromatic vs alkene vs alkyl halide) and
what is the attacker (electrophile vs nucleophile)?'' The four-cell
grid pins the mechanism uniquely.
Concept used. Mechanism categories: EAS (Ar + E+), SNAr
(Ar-X + Nu- with -M activation), electrophilic addition (alkene +
E+), nucleophilic substitution (RX + Nu-), elimination (RX +
strong base).
For each row, identify substrate + attacker.
Map to category.
Matches as above.
Q 6.84
Match the structures given in Column I with the names in Column II. [2pt]
tabularp0.5p0.4
Column I (Structure) & Column II (Name)
(i) CH3-CH=CH-CHBr-CH3 & (a) 4-Bromopent-2-ene
(ii) (CH3)2C(Br)-CH=CH-CH3 & (b) 4-Bromo-3-methylpent-2-ene
(iii) CH3-CH=CH-CH(CH3)-CH2Br? (i.e., CH3CH=C(CH3)CHBr-...) & (c) 1-Bromo-2-methylbut-2-ene
(iv) BrCH2-CH=CH-CH(CH3)2 & (d) 1-Bromo-2-methylpent-2-ene
tabular
Matches. (i)→(a); (ii)→(c); (iii)→(b); (iv)→(d).
Concept used. Apply IUPAC rules: parent chain = longest
chain through the double bond; number from the end nearest the
double bond, then check that the substituent locants are also
minimised.
(i) Pent-2-ene with Br on C4→4-bromopent-2-ene (a).
(ii) But-2-ene with CH2Br at C4, methyl at
C3⇒ but-2-ene with Br on
=CH-CH2Br extension: this fits
1-bromo-2-methylbut-2-ene (c).
(iii) Pent-2-ene parent with Br on C4 and
methyl on C3→4-bromo-3-methylpent-2-ene (b).
(iv) Pent-2-ene parent with Br on C1 and
methyl on C2→1-bromo-2-methylpent-2-ene (d).
(i)→(a); (ii)→(c); (iii)→(b); (iv)→(d).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
IUPAC-walk angle. Trace the longest carbon chain through
the double bond; number from the end nearest to the double bond
(double bond gets lowest locant); cite all substituents alphabetically
with their locants.
Concept used. Lowest-locant rule for principal characteristic
group (the double bond), then for substituents. Substituents cited
in alphabetical order ignoring multiplying prefixes.
For each structure, identify longest chain through C=C.
Number to give C=C the lowest locant.
Identify and locate substituents.
Matches as tabulated.
Q 6.85
Match the reactions given in Column I with the names given in Column II. [2pt]
tabularp0.55p0.38
Column I (Reaction) & Column II (Name)
(i) C6H5-X + R-X + 2Na (ether) →C6H5-R + 2NaX & (a) Fittig reaction
(ii) 2 C6H5-X + 2Na (ether) →C6H5-C6H5 + 2NaX & (b) Wurtz–Fittig reaction
(iii) C6H5-N2+ X- + Cu2X2→C6H5-X + N2 & (c) Finkelstein reaction
(iv) C2H5-Cl + NaI (dry acetone) →C2H5-I + NaCl & (d) Sandmeyer reaction
tabular
Matches. (i)→(b); (ii)→(a); (iii)→(d); (iv)→(c).
Concept used. Each reaction has a canonical name attached:
Finkelstein: R-Cl + NaI -> R-I + NaCl in dry acetone.
(i) Aryl-X + alkyl-X with Na: Wurtz-Fittig (b).
(ii) Two aryl-X with Na: Fittig (a).
(iii) Diazonium + Cu2X2: Sandmeyer (d).
(iv) RCl + NaI in acetone: Finkelstein (c).
(i)→(b); (ii)→(a); (iii)→(d); (iv)→(c).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Pattern-recognition angle. Named reactions are best learned
as ``reagent fingerprints''. Once you can pattern-match the reagent
combination, the name and product follow immediately.
Concept used. The four name reactions here all install a
halogen or couple a C-X to make a new C-C bond. Wurtz/Fittig/Wurtz-Fittig
use Na; Sandmeyer uses Cu(I); Finkelstein uses NaI in dry acetone
to swap halides.
For each row, identify reagent fingerprint.
Map to canonical name.
Matches as above.
V. Assertion and Reason Type
Q 6.86
Assertion (A): Phosphorus chlorides (tri and penta) are preferred over thionyl chloride for the preparation of alkyl chlorides from alcohols. Reason (R): Phosphorus chlorides give pure alkyl halides.
Correct option: (ii) Both A and R are wrong.
Concept used. Thionyl chloride SOCl2 is in fact
preferred over PCl3/PCl5 for alcohol-to-alkyl-chloride
conversion because the by-products (SO2 gas and HCl gas)
both escape, leaving the pure alkyl chloride behind. PCl3
yields H3PO3 (phosphorous acid) as a co-product that
contaminates the product.
Both A (PCl preferred) and R (P-Cl gives pure) are incorrect.
Option (ii): both A and R are wrong.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
By-product-cleanness angle. The deciding factor is what
leaves with the product. Gaseous by-products are removed
automatically; liquid by-products remain and require separation.
Concept used. The Darzens or Hell-Volhard-Zelinsky family of
acid-halide-forming reagents are chosen primarily for clean
work-up. SOCl2 wins because both by-products are gases.
Write the reaction for each reagent.
Identify by-products.
Gases → cleaner; liquids → contaminating.
Option (ii).
Q 6.87
Assertion (A): The boiling points of alkyl halides decrease in the order: RI > RBr > RCl > RF. Reason (R): The boiling points of alkyl chlorides, bromides and iodides are considerably higher than those of the hydrocarbon of comparable mass.
Correct option: (v) Both A and R are correct statements but R is not the correct explanation of A.
Concept used. A is correct: heavier halide ⇒
more polarisable ⇒ stronger dispersion forces
⇒ higher boiling point. R is also correct (alkyl halide
Tb > alkane of same carbon count) but is a separate fact,
comparing halide to hydrocarbon rather than halide-to-halide.
Therefore R doesn't explain A.
Verify A: I (largest, most polarisable) → highest Tb.
Verify R: C2H5Cl (12.3∘C) > ethane (-89∘C).
R compares R-X to R-H, not different halides among themselves.
Conclude R doesn't explain A.
Option (v).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Logical-relation angle. The trick is to ask: does R
provide the causal chain for A? Here R is a separate true
statement that doesn't address halide-vs-halide comparison.
Concept used. For A-R type questions, R must logically
imply A; if A and R are both true but unrelated, the answer is (v).
Test R→A: does ``R-X boils higher than R-H''
imply ``RI boils higher than RBr''? No.
Both A and R are individually correct but unrelated ⇒ (v).
Option (v).
Q 6.88
Assertion (A): KCN reacts with methyl chloride to give methyl isocyanide. Reason (R):CN- is an ambident nucleophile.
Correct option: (iv) A is wrong but R is correct.
Concept used. A is wrong: KCN (ionic, dissociates fully)
delivers CN- free in solution; attack occurs through the
softer, more nucleophilic C end, giving methyl cyanide
(nitrile, CH3-CN), not isocyanide. AgCN (covalent, holds the
C tightly through Ag-C bond) leaves the N end free and gives
isocyanide. R is correct: CN- is indeed ambident.
KCN → free CN-; C-attack →CH3-CN (acetonitrile).
AgCN → Ag-bound C; only N free; N-attack →CH3-NC (isocyanide).
A confuses these two; R is true.
Option (iv).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Reagent-identity angle. The identity of the metal cation
(K+ vs Ag+) controls which end of cyanide is free. Ionic K-CN
releases free anion; covalent Ag-CN ties up the C and exposes only
N.
Concept used. Hard-soft acid-base matching plus electrostatic
binding: Ag+ binds C tightly (soft-soft), K+ doesn't
bind C at all (hard-soft mismatch).
Identify reagent: KCN.
Determine which end is nucleophilic: C end (free CN-).
Product: methyl cyanide (nitrile), not isocyanide.
Option (iv).
Q 6.89
Assertion (A):tert-Butyl bromide undergoes Wurtz reaction to give 2,2,3,3-tetramethylbutane. Reason (R): In Wurtz reaction, alkyl halides react with sodium in dry ether to give hydrocarbon containing double the number of carbon atoms present in the halide.
Correct option: (i) Both A and R are correct and R is the correct explanation of A.
Concept used. Wurtz: 2R-X + 2Na -> R-R + 2NaX. For
R = (CH3)3C-, the dimer is
(CH3)3C-C(CH3)3 = 2,2,3,3-tetramethylbutane. Each
tert-butyl contributes 4 carbons; product has 8.
Count carbons in product: 2 × 4 = 8⇒ R is
the explanation of A.
Option (i).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Coupling-doubling angle. The Wurtz reaction couples two
alkyl halide fragments, doubling the carbon count. Apply directly
to (CH3)3CBr.
Concept used. Wurtz mechanism: Na donates one electron to
R-X to give an alkyl radical; two radicals couple. Practically,
this gives substantial Wurtz product for tertiary halides despite
the tendency to eliminate.
Assertion (A): Presence of a nitro group at ortho or para position increases the reactivity of haloarenes towards nucleophilic substitution. Reason (R): Nitro group, being an electron withdrawing group, decreases the electron density over the benzene ring.
Correct option: (i) Both A and R are correct and R is the correct explanation of A.
Concept used.-NO2 at o/p accepts the negative
charge of the Meisenheimer intermediate by resonance, stabilising
it. The lowered Meisenheimer energy translates to lower activation
energy and faster SNAr. The mechanism is exactly
``NO2 pulls electron density out of the ring''.
A: o/p-NO2 activates Ar-X toward SNAr.
R: -NO2 is electron-withdrawing → ring electron density
drops → Meisenheimer intermediate (negative on ring) is more stable.
R directly explains A.
Option (i).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Resonance-stabilisation angle.NO2 at o/p provides
a direct resonance sink for the negative charge of the Meisenheimer
adduct. R restates the same physics in different words; A and R are
two sides of one coin.
Concept used. SNAr rate enhancement by -M groups at
o/p. Picryl chloride is the canonical maximally-activated example.
Confirm A: o/p-NO2 accelerates SNAr.
Confirm R: NO2 is electron-withdrawing.
R explains A.
Option (i).
Q 6.91
Assertion (A): In monohaloarenes, further electrophilic substitution occurs at ortho and para positions. Reason (R): Halogen atom is a ring deactivator.
Correct option: (v) Both A and R are correct statements but R is not the correct explanation of A.
Concept used. A is correct: halogens are o/p-directing in
EAS. R is correct: halogens are net deactivators (overall slower
than benzene). But R doesn't explain A: deactivation would predict
m-direction (like NO2 or CN); o/p direction is
specifically due to resonance donation from the halogen lone pair,
not from deactivation.
A: halogens direct o/p. True (resonance donation).
R: halogens are deactivating. True (-I > +M on average).
But the reason halogens direct o/p is +M donation,
not deactivation ⇒ R is correct but doesn't explain A.
Option (v).
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Two-effect angle. Halogens carry both -I (withdrawal,
deactivating) and +M (donation, o/p-directing). The two effects
explain different observations: deactivation explains the slower
rate; resonance donation explains the o/p regiochemistry.
Concept used. Substituent effects in EAS: directing
controlled by ± M (resonance, looks for lone pairs/π-acceptor);
activation controlled by ± I + ± M algebraic sum.
Verify both A and R individually.
Test whether R causes A.
Conclude unrelated ⇒ (v).
Option (v).
Q 6.92
Assertion (A): Aryl iodides can be prepared by reaction of arenes with iodine in the presence of an oxidising agent. Reason (R): Oxidising agent oxidises I2 into HI.
Correct option: (iii) A is correct but R is wrong statement.
Concept used. A is correct: iodination of arenes is
reversible, and adding an oxidant pulls the equilibrium toward
Ar-I by destroying the reducing HI by-product. R is wrong: the
oxidant oxidises HI back to I2 (and water), not the
other way around.
Iodination: ArH + I2 <=> Ar-I + HI (slow forward, fast reverse).
Direction-of-oxidation angle. HI is the reductant (low
oxidation state of I); I2 is the oxidised form. Oxidising HI
yields I2 + water. R confuses the direction.
Concept used. Le Chatelier driving an equilibrium. Removing
HI (by oxidising it back to I2) shifts ArH + I2 <=> ArI + HI
to the right.
Establish reversibility of arene iodination.
Identify which side of the oxidation reaction HI is on.
Reject R as written in reverse.
Option (iii).
Q 6.93
Assertion (A): It is difficult to replace chlorine by -OH in chlorobenzene in comparison to that in chloroethane. Reason (R): Chlorine-carbon (C-Cl) bond in chlorobenzene has a partial double bond character due to resonance.
Correct option: (i) Both A and R are correct and R is the correct explanation of A.
Concept used. The C-Cl in chlorobenzene is shorter
and stronger because of resonance donation from Cl's lone
pair into the ring (partial double bond character). The stronger
bond is harder to break, so OH- struggles to displace
Cl-. Industrial conversion of chlorobenzene to phenol (Dow
process) requires 300∘C and 300 atm.
Confirm A: PhCl is far less reactive in SN than EtCl.
Confirm R: PhCl has partial double bond character due to resonance.
R explains A through bond strength.
Option (i).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Bond-strength angle. Stronger C-X bond ⇒ slower
SN. The resonance contribution from Cl's lone pair
adds partial double-bond character, shortening and strengthening
PhCl's C-Cl by ∼ 60 kJ/mol over the alkyl case.
Concept used. Partial double-bond character via resonance
in haloarenes is the single key fact that explains PhCl's
unreactivity in SN. Same logic applies to phenol's
unbreakable C-O.
Verify A.
Verify R.
Confirm R causes A.
Option (i).
Q 6.94
Assertion (A): Hydrolysis of (-)-2-bromooctane proceeds with inversion of configuration. Reason (R): This reaction proceeds through the formation of a carbocation.
Correct option: (iii) A is true but R is false.
Concept used.(-)-2-bromooctane is a secondary
alkyl halide. With OH- (good nucleophile) in protic medium,
it undergoes SN2, not SN1. SN2
proceeds via a single concerted step with back-side
attack, producing complete Walden inversion at the
chiral centre.
Verify A: hydrolysis converts (-)-2-bromooctane to
(+)-2-octanol with inversion of configuration.
(A is true.)
Examine R: ``proceeds through a carbocation'' would be
SN1, which gives racemisation, not
clean inversion. Secondary substrates in basic aqueous
OH- do not generate free carbocations. R is false.
Therefore (iii): A true, R false.
Option (iii): A is true, R is false. Hydrolysis is SN2 (Walden inversion), not via a carbocation.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Stereo-cue angle. Stereochemistry is the surest mechanistic
tell-tale. ``Inversion'' is the unmistakable keyword for
SN2 (Walden inversion through back-side attack);
``racemisation'' is the unmistakable keyword for SN1
(planar cation, both faces equally accessible). Whenever an
assertion mentions clean inversion and the reason invokes a
carbocation, the two are mechanistically incompatible. The
assertion must be true while the reason is false –- option (iii).
Concept used.(-)-2-Bromooctane is a chiral secondary
alkyl halide. With OH- in aqueous medium the rate law is
r = k[R-Br][OH-], indicating bimolecular
SN2. The mechanism is one concerted step: the
nucleophile approaches the carbon from the side opposite the
leaving group; the three remaining substituents pivot through a
trigonal-bipyramidal transition state (sp2-like at the
α-carbon); the chiral centre emerges with its
configuration mirror-imaged –- the celebrated Walden
inversion. The product (+)-2-octanol is the stereo-inverted
enantiomer of the starting material.
Test A. Does (-)-2-bromooctane hydrolyse with
inversion? Experimentally yes –- (+)-2-octanol is
obtained with ≥ 99% inversion. A is true.
Test R. Does the reaction proceed via a
carbocation? A secondary substrate plus a strong
nucleophile (OH-) in a protic but predominantly
bimolecular regime ⇒SN2, not
SN1. No free carbocation forms. R is false.
Reconcile. If R were correct (cation
intermediate) then A could not be (cations racemise,
not invert). Hence A true + R false ⇒ option
(iii).
Option (iii): A is true, R is false. Hydrolysis is SN2 with Walden inversion, not via a carbocation.
Q 6.95
Assertion (A): Nitration of chlorobenzene leads to the formation of m-nitrochlorobenzene. Reason (R):-NO2 group is an m-directing group.
Correct option: (iv) A is false but R is true.
Concept used. The directing effect in EAS is set by the
group already on the ring (here Cl), not by the
incoming group (NO2). Chlorine is weakly deactivating but
ortho/para directing (resonance donation from Cl lone
pair > inductive withdrawal at o/p positions in the
Wheland intermediate). So nitration of chlorobenzene gives mainly
o- and p-nitrochlorobenzene, notm.
Locate the directing group in the substrate: it is
-Cl, an o/p-director.
Predict products of C6H5Cl + HNO3/H2SO4:
o-nitrochlorobenzene (∼ 30%) +p-nitrochlorobenzene
(∼ 70%). m-isomer is minor (∼ 1%).
A claims m-product is major –- false.
R: ``NO2 is m-directing'' –- true when it is
already on the ring (deactivating, m-director). But in
the present reaction NO2 is the incoming, not the
resident, group; it doesn't direct itself.
Hence A false, R true ⇒ option (iv).
Option (iv): A is false, R is true. Chlorine, being o/p-directing, sends NO2 mainly to o and p positions.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Substrate-rules-incoming angle. In electrophilic aromatic
substitution, the regiochemistry is dictated by the group
already on the ring, never by the incoming group. Read
the assertion through that lens: chlorobenzene is the substrate,
so the resident Cl decides where NO2 ends up.
Cl is an o/p-director (deactivating but o/p-directing,
the famous halogen exception), so the major products must be
o- and p-nitrochlorobenzene. Assertion A is therefore false.
Concept used. Substituent directing effects are governed
by the resonance structures of the Wheland intermediate.
-Cl has lone pairs that can donate into the ring; the
resulting positive charge in the Wheland intermediate is best
stabilised when the electrophile attaches at o or p (these
are the positions where the lone-pair donation places the
positive charge on a carbon bearing Cl). The -NO2
group is indeed a m-director, but only when it is already
mounted on the ring; in this question NO2 is the
incoming group, not the resident.
Identify the substrate's directing group.
The ring carries -Cl; nothing else. Cl is
deactivating +o/p-directing.
Predict products.C6H5Cl + HNO3/H2SO4
gives o-nitrochlorobenzene (∼ 30%) and
p-nitrochlorobenzene (∼ 70%). The m-isomer is
a tiny side product (∼ 1%).
Evaluate A. A claims m-nitrochlorobenzene is
the product. That is false; m- is negligible.
Evaluate R. R asserts that -NO2 is a
m-director. As a general statement (when NO2
is already on the ring) this is true.
Match the answer key. A false + R true
⇒ option (iv).
Option (iv): A is false, R is true. Resident Cl on the ring is o/p-directing, so NO2 goes mainly to o and p positions; m- is not the major product.
VI. Long Answer Type
Q 6.96
Some alkyl halides undergo substitution whereas some undergo elimination on treatment with bases. Discuss the structural features of alkyl halides which are responsible for this difference, with examples.
Concept used. The competition between
nucleophilic substitution (SN1/SN2)
and β-elimination (E1/E2)
depends on (a) class of the alkyl halide (1∘, 2∘,
3∘), (b) strength and bulk of the base/nucleophile, (c)
temperature, and (d) solvent. We tabulate the dominant behaviour.
Primary R-CH2-X: preferred path is
SN2 with a strong nucleophile in protic solvent
(e.g. CH3CH2-Br + OH-/H2O -> CH3CH2-OH). With a
strong, bulky base ((CH3)3CO- K+, ``t-BuOK'')
the path flips to E2 giving the alkene.
Secondary R2CH-X: sits on the fence. With
OH-/water → mixed SN2/E2;
with alc. KOH (hot, less polar) → predominantly
E2. Example: CH3-CHBr-CH3 + alc. KOH ->
CH2=CHCH3 (propene).
Tertiary R3C-X: cannot do SN2
(sterically blocked). With weak base/protic solvent →SN1 (ionisation, racemic product). With strong
base or high temperature →E1/E2
(Saytzeff alkene). Example: (CH3)3C-Br + alc. KOH ->
(CH3)2C=CH2 (isobutene).
Effect of base: small, charge-localised, basic
(OEt-, OH- in alcohol) favours
E2. Big polarisable nucleophiles (I-,
RS-) favour SN2.
Effect of solvent and temperature: polar protic
+ low T favours substitution; polar aprotic + high
T favours elimination (and changes selectivity by an
order of magnitude).
Class of R-X + nature/bulk of base + temperature decide substitution vs elimination. 1∘→ SN2, 2∘→ mixed, 3∘→ E1/SN1. Bulky strong base (t-BuOK) routes any class to E2.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Decision-tree angle. Three structural and experimental
questions are sufficient to predict whether substitution or
elimination will dominate:
2pt
Is the substrate 1∘, 2∘ or 3∘?
Is the base/nucleophile small or bulky, weak or strong?
Is the reaction run cold (room temperature) or hot
(reflux in alcohol)?
Combine the three answers via the decision table given in the
SOLUTION and you have the dominant pathway in 15 seconds, which
is faster than re-deriving the mechanism from scratch on the
exam.
Concept used. Substitution requires nucleophilic
approach to the α-carbon; elimination requires base
abstraction of a β-hydrogen followed by departure of the
leaving group. Bulky bases (t-BuOK, LDA) cannot reach the
α-carbon (sterically blocked) but they can pluck
off a β-H, so they push the reaction toward E2.
Polar protic solvents at low T favour ionisation
(SN1); polar aprotic solvents at high T favour
bimolecular E2 or SN2 (the entropy term
TΔ S in E2 becomes important at high T,
since elimination produces two molecules from one).
Worked example A:(CH3)3C-Cl + NaOEt
in EtOH at 25∘C. Tertiary substrate + small
base + polar protic solvent at moderate T⇒E1 (Saytzeff isobutene major,
∼ 90%) with minor SN1 ether by-product
(∼ 10%).
Worked example B: Switch the base to
(CH3)3CO-K+ (t-BuOK), keep substrate and
solvent. Bulky base cannot reach the α-C of
a 3∘ carbon, but readily plucks the β-H.
Result: E2 exclusively, 100% isobutene.
Worked example C:CH3CH2-Br + NaI in
acetone. Primary substrate + soft polarisable
nucleophile + polar aprotic solvent ⇒
pure SN2 (Finkelstein); no elimination
because I- is a poor base.
Decision-rule recap. ``Substrate first,
base second, temperature third.'' Tertiary in alc. KOH
⇒ E2; primary in NaI/acetone ⇒
SN2; secondary in H2O/OH-⇒ mixed (often ∼ 60:40).
Substrate class + base nature/bulk + temperature jointly determine substitution vs elimination. 1∘→ SN2; 3∘→ E1/SN1; bulky strong base (t-BuOK) always routes the reaction to E2.
Q 6.97
Some halogen containing compounds are useful in daily life. Some compounds of this class are responsible for exposure of flora and fauna to more and more of UV light which causes destruction to a great extent. Name the class of these halocompounds. In your opinion, what should be done to minimise harmful effects of these compounds?
Concept used. The compounds that destroy stratospheric
ozone (and so increase UV exposure at Earth's surface) are the
chlorofluorocarbons (CFCs), also called Freons.
Representative examples are CCl3F (CFC-11), CCl2F2
(CFC-12, Freon-12), and C2Cl3F3 (CFC-113). They are
unreactive at ground level (which is why they were popular as
refrigerants, aerosol propellants, foam-blowing agents and
solvents) but in the stratosphere they are split by UV radiation
into Cl radicals, which catalyse the destruction of ozone:
CCl2F2 ->[hν] CClF2• + Cl•
Cl• + O3 -> ClO• + O2
ClO• + O3 -> Cl• + 2O2
A single Cl radical can destroy ∼ 105 ozone molecules
before it is sequestered (typically as HCl or ClONO2).
The resulting thinning of the ozone layer (especially the
``Antarctic ozone hole'') allows more UV-B radiation to reach the
surface, causing skin cancer, cataracts, immune suppression in
humans, and damage to phytoplankton and crop plants.
Steps to minimise harmful effects:
Phase-out CFCs. Replace them with
hydrofluorocarbons (HFCs, e.g. R-134a,
CH2F-CF3) and hydrochlorofluorocarbons (HCFCs)
that decompose lower in the atmosphere and don't reach the
stratosphere intact. HFCs contain no Cl, so even if
they reach the stratosphere they don't catalyse ozone loss.
Enforce international agreements. The
Montreal Protocol (1987, with subsequent amendments)
has phased out CFCs worldwide; the Kigali Amendment (2016)
adds HFCs to the phase-down list because they are potent
greenhouse gases.
Recover and recycle. Old refrigerators, ACs and
fire extinguishers must be recovered carefully to prevent
CFC leakage; mandatory return programmes help.
Develop greener alternatives.
Hydrocarbon refrigerants (isobutane, propane) and natural
refrigerants (NH3, CO2) have zero ozone
depletion potential and very low global-warming potential.
Public-awareness measures. Encourage purchase of
CFC/HFC-free products; reduce reliance on aerosol cans;
increase shade and SPF use to reduce UV exposure damage.
The Antarctic ozone hole, first reported in 1985, has stabilised
since the 1990s thanks to the Montreal Protocol and is projected to
recover by ∼ 2065 to pre-1980 levels.
CFCs (chlorofluorocarbons, Freons) destroy the ozone layer. Mitigation: phase out CFCs (Montreal Protocol), substitute with HFCs/HCFCs/hydrocarbons, recover-and-recycle, develop greener alternatives.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Molecule-mechanism-policy angle. CFCs are the classic case
where a useful synthetic chemical has unintended consequences far
from the application site. The chain of cause is:
(1) CFC inertness ⇒ years-long stratospheric
lifetime; (2) UV photolysis releases Cl.;
(3) Cl-O catalytic cycle depletes ozone;
(4) thinned ozone ⇒ more UV-B at the surface;
(5) biological damage (DNA damage, cataracts, ecosystem stress).
Concept used. Atmospheric chemistry: tropospheric
inertness lets CFCs reach the stratosphere; once there, the
high-energy UV cleaves the otherwise robust C-Cl bond. The chlorine
atom is a catalyst, not a stoichiometric consumer, so a small mass
of CFC can destroy a huge mass of ozone over its multi-decade
stratospheric residence.
Quantify the damage. One Cl atom destroys
∼ 105 O3 molecules before deactivation.
Plan the response. Phase-out, substitution,
recovery, public awareness; backstopped by international
regulation (Montreal Protocol).
Track progress. Atmospheric CFC concentrations
peaked in ∼ 1994 and are now slowly falling; ozone
layer recovery is on track.
CFCs are the class; mitigation: international phase-out, safer substitutes, recovery and recycling.
Q 6.98
Why are aryl halides less reactive towards nucleophilic substitution reactions than alkyl halides? How can we enhance the reactivity of aryl halides?
Concept used. The unreactivity of aryl halides combines
three structural effects; reactivity is boosted by electron
withdrawal from the ring.
Resonance shortening. In chlorobenzene, Cl's
lone pair donates into the ring, giving C-Cl a
partial double-bond character. Bond length ∼ 169 pm
vs ∼ 178 pm in CH3Cl; the shorter, stronger
bond is harder to break.
Hybridisation. The aryl C is sp2
(33% s), the alkyl C is sp3 (25% s). The
sp2-X bond is stronger and shorter because s
electrons sit closer to the nucleus.
Repulsive transition state. An aryl cation
(SN1) would be a destabilised sp2 ion with
the empty orbital in the ring plane –- not in conjugation
with the π-system. SN2 back-side attack is
blocked by the ring's π-cloud and by the geometry of
the ipso carbon.
Activation toward SNAr. Strong
electron-withdrawing groups (especially -NO2) at
o- and p-positions stabilise the
Meisenheimer intermediate by delocalising the
negative charge onto the nitro oxygens:
Cl-C6H3(NO2)2 + OH- -> [Meisenheimer-]
-> HO-C6H3(NO2)2 + Cl-.
2,4-Dinitrochlorobenzene reacts with aqueous NaOH
at ∼ 100∘C; 2,4,6-trinitrochlorobenzene
(picryl chloride) reacts at room temperature.
Industrial route (Dow process). Even
unactivated chlorobenzene reacts with NaOH at
300∘C and ∼ 300 atm to give phenol.
Severe conditions are needed precisely because the
substrate is so unreactive.
Position matters.m-nitro substituents do not
stabilise the Meisenheimer adduct (resonance only reaches
o and p positions), so m-nitrochlorobenzene is barely
more reactive than chlorobenzene itself.
Aryl halides are sluggish in SN because of resonance shortening of C-X, sp2 hybridisation, and unstable aryl cation/blocked SN2 TS. Reactivity is enhanced by introducing -NO2 (or other strong -M) groups at o- and p-positions, which stabilise the Meisenheimer intermediate via the SNAr pathway.
VP
Vivaan Patel
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Resonance-stabilisation angle. The trick to making an
otherwise inert aryl halide reactive is to provide a structural
mechanism that stabilises the negative charge which transiently
develops on the ring during nucleophilic attack
(SNAr). -NO2 groups at o/p accept that
charge through resonance into their formal N=O bonds.
Without such activation, the ring would have to host a localised
carbanion lobe on a particular ring carbon, an extremely
high-energy species; with NO2 activation at o or p,
the carbanion lobe delocalises directly onto the nitro oxygens
where the negative charge is comfortable.
Concept used. The two-step addition–elimination
mechanism (SNAr, sometimes called the
Meisenheimer mechanism) is the only viable pathway for
nucleophilic substitution on aryl halides. It requires the
substrate to host a strong -M (electron-withdrawing by
resonance) group whose lone-pair-accepting orbital can reach the
ipso carbon through the ring's π-framework. Only o and p
substituents satisfy this geometric requirement; m-substituents
sit at the wrong nodes of the carbanion's molecular orbital and
provide no stabilisation. That is why m-nitrochlorobenzene is
barely more reactive than chlorobenzene itself.
Diagnose unreactivity. Three structural defences
sustain it: resonance shortening of C-X (partial
double-bond character, ∼ 169 pm vs ∼ 178 pm in
CH3Cl), sp2 hybridisation at the ipso carbon
(stronger bond), and a transition state that
destabilises both SN1 (aryl cation has empty
orbital in the ring plane) and SN2 (the
π-cloud blocks back-side attack).
Plan the activation. Add -NO2 at o
and/or p. The Meisenheimer intermediate now has
resonance structures placing (-) on the nitro
oxygens, dropping its energy by tens of kJ/mol.
Quantify the rate gain. Chlorobenzene needs
300∘C and 300 atm (Dow process) to give
phenol with NaOH. 2,4-Dinitrochlorobenzene reacts
with aqueous NaOH at ∼ 100∘C and
1 atm. 2,4,6-Trinitrochlorobenzene (picryl chloride)
reacts with H2O at room temperature –- a ∼
1012 rate boost over chlorobenzene.
Industrial leverage. The SNAr
activation strategy underpins the synthesis of
sulphonamide drugs, herbicides, and azo dye
intermediates, all built from o/p-nitrohaloarenes.
Aryl halides are unreactive in classical SN1/SN2 because of resonance, hybridisation and TS effects. They react via SNAr only when activated by -NO2 (or similar -M) groups at o/p positions, which stabilise the Meisenheimer intermediate by resonance.
More Haloalkanes and Haloarenes Class 12 Chemistry Resources
Collegedunia hosts six sibling resources for this chapter, each canonical for one role.
Haloalkanes and Haloarenes Class 12 Chemistry Exemplar Solutions FAQs
Q. How many problems are there in the Class 12 Chemistry Chapter 6 Haloalkanes and Haloarenes Exemplar?
The Haloalkanes and Haloarenes Exemplar has 80 problems across MCQ-I (30), MCQ-II (10), Short Answer (20), Matching (3) and Assertion-Reason / LA (17). The Collegedunia PDF works 25 representative items covering every type.
Q. Is the Haloalkanes and Haloarenes chapter Chapter 6 or Chapter 10 in NCERT?
Under the current 2026-27 NCERT, Haloalkanes and Haloarenes is Chapter 6 of Class 12 Chemistry. Older prints and many third-party sites still list it as Chapter 10, but the content is unchanged.
Q. What is the CBSE weightage of Haloalkanes and Haloarenes in the Class 12 board exam?
The chapter carries roughly 6 to 8 marks, usually as one 3-mark SA on SN1 or SN2 mechanism, one 2-mark VSA on a named reaction (Sandmeyer, Finkelstein, Wurtz-Fittig), and a 5-mark LA on chirality and stereochemistry in alternate years.
Q. Which topics from Haloalkanes and Haloarenes are most important for JEE Main and NEET?
The highest-yield topics are SN1 vs SN2 prediction, carbocation stability ranking, stereochemistry (inversion, retention, racemisation), Saytzeff elimination, and the named reactions Sandmeyer, Finkelstein, Wurtz-Fittig and Fittig.
Q. Are the Exemplar problems harder than the NCERT textbook exercises?
Yes. The Exemplar reframes textbook facts as multi-factor reactivity rankings, asks for comparisons across two substrates, and tests assertion-reason logic on stereochemistry. The Collegedunia Exemplar Solutions PDF works each item with a Solution plus an Expert's Solution that names the controlling factor.
Q. Can I rely only on Exemplar Solutions for board exam prep on Haloalkanes and Haloarenes?
The Exemplar covers the conceptual depth a board exam tests, but pair it with the NCERT textbook back-exercises for completeness on standard mechanism-drawing and named-reaction questions.
Q. How do I download the Haloalkanes and Haloarenes Exemplar Solutions PDF for free?
Use the download button at the top of this page to get the free PDF of NCERT Exemplar Solutions for Class 12 Chemistry Chapter 6 Haloalkanes and Haloarenes, fully aligned to the 2026-27 syllabus.
Q. What is the difference between Saytzeff and Hofmann rules in Exemplar elimination problems?
Saytzeff's rule states that with a small base (alc. KOH, ethoxide) the major alkene in β-elimination is the more substituted (more stable) one. Hofmann's rule applies with bulky bases (potassium tert-butoxide): the less-substituted (less hindered) alkene becomes the major product. The Exemplar tests this every alternate year as an MCQ-II or matching item.
Q. What is the Kharasch (peroxide) effect and why does it apply only to HBr?
The Kharasch peroxide effect is the anti-Markovnikov addition of HBr to an unsymmetrical alkene in the presence of organic peroxides (R-O-O-R). The Br radical attaches to the less-substituted carbon. The effect operates only for HBr because only the Br radical chain is energetically favourable; H-F, H-Cl and H-I either have too strong or too weak X-H / C-X bonds for the chain to propagate.
Q. How are R and S configurations assigned to a chiral haloalkane?
Rank the four substituents on the chiral carbon by CIP priority (highest atomic number = 1). Orient the molecule so the lowest-priority group points away from you. Trace 1 to 2 to 3: clockwise gives R (rectus); anti-clockwise gives S (sinister). Exemplar MCQ-II often asks the R/S of 2-bromobutane after SN1 (racemic) or SN2 (inverted) substitution.
Q. What is the role of anhydrous ZnCl2 in the Lucas test for alcohols?
Anhydrous ZnCl2 + conc. HCl is the Lucas reagent for distinguishing primary, secondary, and tertiary alcohols by their rate of conversion to the alkyl chloride. ZnCl2 acts as a Lewis-acid catalyst that polarises the C-O bond. Tertiary alcohols turn cloudy immediately; secondary in 5-10 minutes; primary, not at room temperature.
Q. How are DDT and freons (CFCs) prepared and why are they environmentally hazardous?
DDT is prepared by condensing chlorobenzene with chloral (CCl3CHO) in conc. H2SO4. Freons (CCl2F2) are made from CCl4 by the Swarts reaction with SbF3/HF or Hg2F2. DDT bio-accumulates in fat tissues along the food chain; CFCs migrate to the stratosphere where UV releases Cl radicals that catalytically destroy ozone. DDT is banned in most countries; CFCs are phased out under the Montreal Protocol.
Q. What is racemisation and why does the SN1 mechanism produce a racemic mixture?
Racemisation is the conversion of an optically active substrate into a 1:1 mixture of (R) and (S) enantiomers (the racemic mixture), which is optically inactive overall. SN1 first ionises the C-X bond to give a planar sp2 carbocation. The nucleophile attacks this planar intermediate from either face with equal probability, producing equal amounts of (R) and (S) product and net zero optical rotation.





















































































































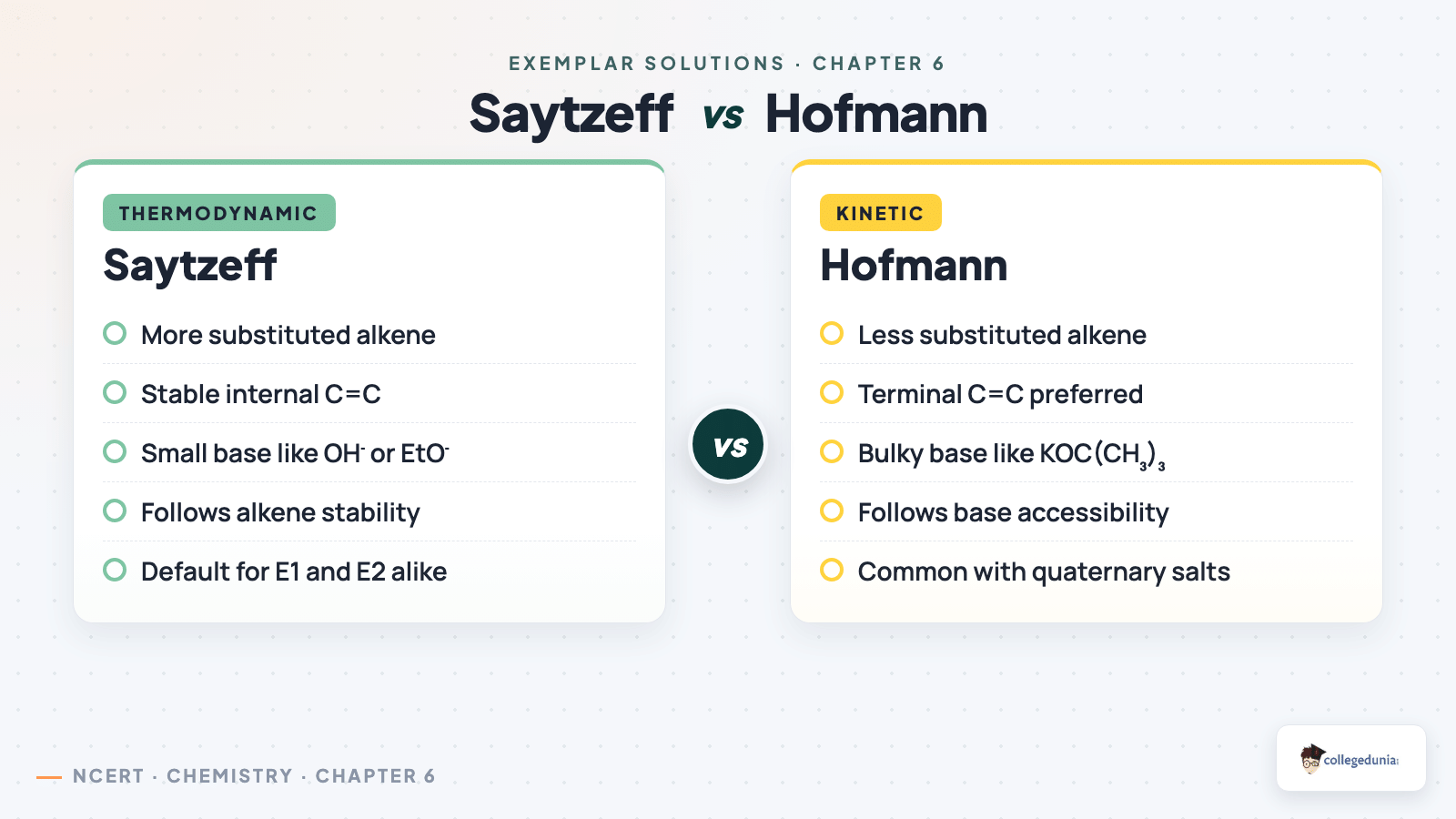


Comments