Chemistry Mentor | B.Tech Student, IIT Delhi | Updated on - May 25, 2026
The Class 12 Chemistry Chapter 3 Chemical Kinetics Exemplar packs 36 problems spread across MCQ-I, MCQ-II, VSA, SA and LA, each pitched at entrance-grade reasoning on rate law, order, half-life, the Arrhenius equation, collision theory, and pseudo-first-order kinetics. The chapter remains intact in the 2026-27 NCERT. This page hosts the worked Exemplar solutions PDF for free download.
The PDF works through all 36 problems with a Solution and a separate Expert's Solution that names each rule invoked.
These Exemplar Solutions are curated by subject experts at Collegedunia, mapped to the 2026-27 NCERT, and benchmarked against the last five years of CBSE Board, JEE Main and NEET papers.
Why the Chemical Kinetics NCERT Exemplar Matters for JEE Main and NEET 2026 Prep
Chemical Kinetics is one of the highest-yield Physical Chemistry chapters for entrance exams because the same problem types recur with a small twist every year. The Exemplar trains exactly the twist: it reframes rate-law data, hides the order inside half-life ratios, and asks for activation energy under a temperature shift.
Across the last five JEE Main shifts, Chemical Kinetics contributed at least one question per shift, three of them direct scaffolds of Exemplar 3.14, 3.18 and 3.32. For NEET, the chapter holds 2 to 3 questions per paper, mostly on the integrated first-order equation and the Arrhenius plot.
Why bother: The Exemplar trains a habit the textbook does not, identifying reaction order from indirect evidence like half-life ratios or pressure data. This is the most common JEE Main Physical Chemistry trap.
How will Collegedunia's NCERT Exemplar Solutions Help You with Chemical Kinetics?
Each of the 36 problems is solved twice: a clean Solution and an Expert's Solution that names every rate law and approximation used.
Every Question Type Worked End-to-End: MCQ-I, MCQ-II, VSA, SA and LA, each with full reasoning.
Concept Stack Named: Differential rate law, integrated equation, half-life, Arrhenius, or pseudo-first-order, called out per step.
JEE and NEET Bridge: Items 3.14, 3.18, 3.27 and 3.32 are tagged with the year that reused their scaffold.
2026-27 Aligned: All 36 problems sit inside the current syllabus; nothing was trimmed.
Best Way to Use the Chemical Kinetics Exemplar for JEE and NEET Prep
A time-boxed pass keyed to question type works better than reading all 36 problems back-to-back. First-pass budget for a student two weeks before a JEE Main attempt:
Question Type
Items
Time per Problem
Total Budget
MCQ-I (single correct)
3.1 to 3.5
2 to 3 min
~13 min
MCQ-II (multiple correct)
3.6 to 3.14
4 to 5 min
~40 min
VSA (1 to 2 marks)
3.15 to 3.22
3 to 4 min
~28 min
SA (3 marks)
3.23 to 3.31
6 to 8 min
~60 min
LA (5 marks)
3.32 to 3.36
10 to 14 min
~60 min
Quick Tip: JEE Main aspirants should clear all 5 MCQ-I and 9 MCQ-II first (they map directly to JEE shifts). NEET aspirants should prioritise MCQ-I and the 8 VSA items, then the LAs on activation energy.
Chemical Kinetics Exemplar Question-Type Tour with One Sample Solved per Type
One reasoned sample per type below; the worked set for all 36 problems sits in the PDF.
For order n, k has units mol1-n Ln-1 s-1. For n = 2 : L mol-1 s-1. Answer: (b).
VSA Sample, Exemplar 3.18 (Pseudo-First-Order in Ester Hydrolysis)
Acid-catalysed hydrolysis of ethyl acetate is bimolecular but behaves first-order because water sits in large excess. r = k[ester][H2O] collapses to r = k'[ester] , with k' = k[H2O] .
SA Sample, Exemplar 3.27 (Half-Life from Concentration-Time Data)
Using ln([A]0 / [A]) = kt with [A] dropping from 0.8 to 0.4 mol L-1 in 15 min: ln 2 = 15k , so k = 0.0462 min-1 and t1/2 = 15 min.
LA Sample, Exemplar 3.32 (Activation Energy from Two Temperatures)
Rate constant doubles from 300 K to 310 K. Two-point Arrhenius:
Plugging ln 2 , R = 8.314 J K-1 mol-1 yields Ea ≈ 53.6 kJ mol-1. The 10 form is in the PDF.
Remember: "Rate doubles for a 10 K rise" gives Ea ≈ 50 to 55 kJ mol-1 near room temperature, a useful numerical sanity-check.
Chemical Kinetics Exemplar MCQ-II Solved: Multiple-Correct Walk-Through
MCQ-II is the most-failed Exemplar type because students fixate on the first option that seems right and stop reading. The verification habit on Exemplar 3.11 is the cure.
Exemplar 3.11. The rate of a first-order reaction depends on (a) reactant concentration (b) product concentration (c) time (d) temperature
(a) True. r = k[A] , so rate is proportional to [A] . Selected.
(b) False. Product concentration is absent from a forward-only first-order rate law.
(c) Time itself does not appear in the rate law; rate changes only because [A] decreases.
(d) True. k is temperature-dependent through Arrhenius. Selected. Answers: (a) and (d).
This same setup reappeared as JEE Main 2024 Session 1 MCQ-II and as NEET 2023 Q42 with the product-concentration distractor untouched.
Watch Out: "Rate depends on time" is the most-picked wrong option in MCQ-II 3.11. Time is implicit through [A](t) , not explicit in the rate law.
Chemical Kinetics Exemplar Assertion-Reason Sample Solved
Assertion-Reason items on Chemical Kinetics recur in CBSE Board Set 56/4/1 and in JEE Main. Use the four-option scheme: both true with reason explaining assertion (A), both true but reason does not explain (B), assertion true reason false (C), assertion false (D).
Assertion. The order of a reaction can be a fraction.
Reason. Order is determined experimentally from the rate law and need not match the stoichiometric coefficients of the balanced equation.
Answer: (A). Both true; the reason is exactly why fractional orders such as 12, 32 and even 0 occur. Acetaldehyde decomposition follows r = k[CH3CHO]3/2, the textbook example. This is the insight Exemplar 3.20 and 3.26 both test.
Chemical Kinetics Class 12th: Difficulty Step-Up from NCERT Textbook to Exemplar
The textbook stays close to worked examples; the Exemplar adds a constraint, inverts the question, or asks for a limit case. Five direct comparisons:
Concept
NCERT Textbook Style
Exemplar Twist
Rate law and order
State the rate law for a reaction
Deduce order from half-life ratios at two [A]0 (3.26)
Integrated first-order
Find k from one [A]0, [A]t, t set
Solve for time when [A] reaches 1/8 of initial (3.29)
Arrhenius equation
Given Ea, T, find k
Given k doubles for a 10 K rise, find Ea (3.32)
Half-life
Quote t1/2 = 0.693 / k
Identify order from t1/2 ∝ 1 / [A]0n-1 (3.30)
Catalyst and Ea
State catalyst lowers Ea
Compute rate ratio with vs without catalyst (3.34)
Exemplar-Specific Common Mistakes in Chemical Kinetics
These slip-ups are specific to the Exemplar's HOTS scaffold and differ from textbook-side mistakes:
Confusing molecularity with order in 3.16 and 3.20: molecularity counts an elementary step, order is the experimental exponent. This phrasing trap cost JEE Main 2024 Session 2 candidates 4 marks in one shift.
Mixing log bases in 3.27 and 3.32: Arrhenius uses ln , textbook tables list 10. Mixing introduces a 2.303 error.
Treating the pseudo-first-order constant as the true bimolecular constant in 3.18: k' = k [H2O] , not k.
Forgetting that zero-order half-life depends on [A]0 in 3.30: t1/2 = [A]0 / (2k) for n = 0 , 0.693 / k for n = 1 , 1 / (k [A]0) for n = 2 .
Skipping the kelvin conversion in 3.32 and 3.33. This single oversight is the most-asked Exemplar idea in CBSE Board sets between 2022 and 2025.
How Frequently Has Chemical Kinetics Been Asked in CBSE, JEE and NEET (Top 3 Recurring Topics)
Three Exemplar topics recur disproportionately across the last five years of board and entrance papers.
Topic
Exemplar Item
Recurrence (last 5 years)
Activation energy from two temperatures (Arrhenius)
3.32, 3.33
3 JEE Main + 2 CBSE Board
Half-life and order identification
3.27, 3.30
2 CBSE Board + 2 NEET
Pseudo-first-order in ester hydrolysis and sugar inversion
Topics Covered in Class 12 Chemistry Chapter 3 Chemical Kinetics Exemplar Solutions
The 36 worked Exemplar problems answer every high-search-volume sub-topic students raise before JEE Main, NEET and the CBSE Board. Use the list as a topic-to-Exemplar-item map.
Rate of reaction class 12: Exemplar 3.1, 3.15 - rate expression from stoichiometry, units, and signs.
Order of reaction vs molecularity: Exemplar 3.16, 3.20 - empirical vs theoretical, fractional vs whole.
First order reaction half life formula: Exemplar 3.27 - t1/2 = 0.693/k, independent of [A]0.
Second order reaction integrated rate law: Exemplar 3.30 - 1/[A] - 1/[A]0 = kt , L mol-1 s-1.
Arrhenius equation derivation: Exemplar 3.32, 3.33 - two-temperature form from k = A e-Ea/RT.
Activation energy graph: Exemplar 3.34 - PE profile with and without catalyst overlay.
Pseudo first order reaction: Exemplar 3.18, 3.21 - ester hydrolysis and inversion of cane sugar.
All NCERT Exemplar Questions for Chemical Kinetics with Step-by-Step Solutions
Every question of the NCERT Exemplar set for Class 12 Chemistry Chapter 3 Chemical Kinetics is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
I. Multiple Choice Questions (Type-I)
Q 3.1
The role of a catalyst is to change 2.2cm0.4pt.
(i) gibbs energy of reaction.
(ii) enthalpy of reaction.
(iii) activation energy of reaction.
(iv) equilibrium constant.
Correct option: (iii) activation energy of reaction.
Concept used. A catalyst is a substance that
increases the rate of a chemical reaction without itself being
consumed. It works by providing an alternative reaction
pathway of lower activation energyEa: the energy
barrier between reactants and the transition state (activated
complex). A catalyst does not change the energies of the reactants
or the products themselves, so it cannot change any state function
of the reaction.
Recall the thermodynamic identities for a reaction at constant
T and p:
Δ G = Gproducts - Greactants,
Δ H = Hproducts - Hreactants.
Both are differences between the products' and the reactants'
state functions. A catalyst does not alter either state.
The equilibrium constant is fixed by Δ G∘:
Δ G∘ = -RT ln Keq.
Since Δ G∘ is unchanged, Keq is
unchanged. So options (i), (ii) and (iv) are eliminated.
Only the activation energy can be lowered. With Ea smaller,
the Arrhenius rate constant
k = A e-Ea/RT grows because the exponent becomes less
negative.
Mental picture
Picture the energy profile as a hill between reactants and products.
A catalyst carves a lower pass through the same hill, leaving the two
valleys (reactants, products) at exactly the same height.
Option (iii): a catalyst changes the activation
energy of the reaction.
PS
Pranav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Energy-profile angle. A clean way to remember this is to
sketch the reaction-energy diagram and ask: ``which segment does the
catalyst touch?''.
Concept used. Activation energy Ea is the vertical
height from the reactant level to the peak of the activated complex.
Thermodynamic quantities (Δ H, Δ G, Keq)
depend only on the reactant and product levels; the path between them
is kinetic, not thermodynamic.
Look at the four options as three thermodynamic quantities
(i, ii, iv) and one kinetic quantity (iii). A catalyst
is a kinetic object; it changes only how fast the
reaction goes, not how far.
Confirm with the Arrhenius equation
k = A e-Ea/RT: drop Ea by, say,
20 kJ/mol at T=300 K and
kcatk = e20000/(8.314 × 300)
= e8.02 ≈ 3.0 × 103,
a ∼ 3000-fold rate enhancement with the same Δ H
and same Keq.
Cross-check: the IUPAC definition of a catalyst explicitly
says it provides a lower-Ea pathway and is regenerated.
Options (i), (ii), (iv) would all violate the
``regenerated/unchanged thermodynamics'' clause.
Common Pitfall
Common pitfall. Students sometimes claim a catalyst
``reduces Δ H''. It does not; it only reduces the height of
the activation barrier above the reactants.
Concept Linkage
Concept linkage. This is the kinetic counterpart of the
thermodynamic statement that Keq depends only on T:
K is set by Δ G∘, k is set by Ea, and a
catalyst affects only the second.
Option (iii): only the activation energy is
lowered by a catalyst.
Q 3.2
In the presence of a catalyst, the heat evolved or absorbed
during the reaction 2.2cm0.4pt.
(i) increases.
(ii) decreases.
(iii) remains unchanged.
(iv) may increase or decrease.
Correct option: (iii) remains unchanged.
Concept used. The ``heat evolved or absorbed'' at constant
pressure is the enthalpy change of the reaction,
Δ Hrxn = Hproducts - Hreactants.
Enthalpy is a state function: its value depends only on the
initial (reactants) and final (products) states, not on the path
between them. A catalyst changes only the path, never the endpoints.
Write the energy profile with and without catalyst. Both
start at Hreactants and both end at
Hproducts. So the vertical gap
Δ H = Hproducts - Hreactants
is the same in both pictures.
Hess's law confirms this: any sequence of steps that takes
the same reactants to the same products gives the same
Δ H. The catalysed pathway is just a different
sequence of elementary steps, so its overall Δ H
matches the uncatalysed one.
Eliminate (i), (ii), (iv): all three claim a change in
Δ H, which violates Hess's law.
Option (iii): the heat exchanged is unchanged
because Δ H is a state function.
AM
Aditi Mehta
M.Sc Physical Chemistry, IIT Madras
Verified Expert
State-function angle. The fastest way to settle questions
about whether ``X is changed by a catalyst'' is to ask: is X a state
function or a path quantity?
Concept used. State functions (U, H, G, S,
Keq) depend only on the start and end states. Path
quantities like Ea depend on the route.
Tabulate the options. (i) and (ii) make heat exchanged
a non-state quantity, which is wrong at constant pressure.
(iv) hedges in the same direction.
Numerical cross-check: combustion of methane releases
Δ H = -890 kJ/mol whether or not platinum
wires (a catalyst) speed up the reaction. The wires only
let the same 890 kJ come out faster.
State the result cleanly: enthalpy in = enthalpy out,
independent of the road taken.
Exam tip. JEE Main 2023 (April shift) recycled this exact
distractor set. Lock in: ``catalyst ⇒ kinetics only''.
Concept Linkage
Concept linkage. ``Heat exchanged'' at constant pressure
is the enthalpy change. Enthalpy is a state function set by the
endpoints; catalysts touch only the path. Same logic kills options
(i), (ii) and (iv).
Option (iii): Δ H is unchanged by a
catalyst.
Q 3.3
Activation energy of a chemical reaction can be determined
by 3cm0.4pt.
(i) determining the rate constant at standard temperature.
(ii) determining the rate constants at two temperatures.
(iii) determining probability of collision.
(iv) using catalyst.
Correct option: (ii) determining the rate constants at two
temperatures.
Concept used. The Arrhenius equationk = A e-Ea/RT links the rate constant k to absolute
temperature T through the activation energy Ea and the
pre-exponential factor A. Taking natural logarithm:
ln k = ln A - EaRT.
For two different temperatures T1 and T2 with rate
constants k1 and k2:
ln(k2k1)
= EaR(1T1 - 1T2).
One unknown (Ea) cannot be extracted from a single
equation ln k1 = ln A - Ea/(R T1) because A
is also unknown. Two equations are needed: option (i) gives
only one, so it is insufficient.
Two rate-constant measurements at T1 and T2 give
two equations. Subtracting them eliminates A and isolates
Ea, hence option (ii) works.
Option (iii) (``probability of collision'') corresponds to
the steric factor P inside the pre-exponential A, not to
Ea. Option (iv) (using a catalyst) changes Ea
to a different value Ea', so it cannot measure the
original Ea.
Option (ii): measure k at two temperatures and
solve for Ea.
AI
Aanya Iyer
Ph.D Physical Chemistry, IISc Bangalore
Verified Expert
Graphical angle. Treat the Arrhenius equation as a straight
line in ln k vs 1/T coordinates. Determining Ea is the
same as measuring the line's slope.
Concept used. ln k = -EaRslope·1T
+ ln Aintercept.
Slope is the ratio Δ(ln k)/Δ(1/T); you need at least two
(1/T, ln k) pairs to evaluate Δ.
One measurement gives a single (1/T, ln k) pair, which is
just a point. A point does not have a slope.
Two measurements at T1, T2 give two pairs; the slope
is
slope =
ln k2 - ln k1(1/T2) - (1/T1),
and Ea = -R slope.
Worked numerical check. If k1 = 1.0 × 10-3 at
T1 = 300 K and k2 = 4.0 × 10-3 at
T2 = 320 K,
ln(k2/k1) = ln 4 = 1.386, 1T1 - 1T2
= 1300-1320
= 2.083× 10-4 K-1, Ea = R1.3862.083× 10-4
= 8.314 × 6655
= 5.53× 104J/mol
≈ 55.3 kJ/mol.
Exam tip. NEET 2024 supplied two (T, k) data points and
asked for Ea in kJ mol-1. Always reach for the
two-point form first.
Concept Linkage
Concept linkage. The slope of ln k vs 1/T is the
standard way industry and research labs report Ea for catalytic
reactors, atmospheric chemistry models, and pharmaceutical shelf-life
predictions.
Option (ii): two rate-constant measurements at
two temperatures pin down Ea.
Q 3.4
Consider Fig. 4.1 and mark the correct option.
(i) Activation energy of forward reaction is E1+E2 and product
is less stable than reactant.
(ii) Activation energy of forward reaction is E1+E2 and
product is more stable than reactant.
(iii) Activation energy of both forward and backward reaction is
E1+E2 and reactant is more stable than product.
(iv) Activation energy of backward reaction is E1 and product is
more stable than reactant.
Fig. 4.1, NCERT Exemplar Class 12 Chemistry, Chapter 4.
Correct option: (i) activation energy of forward reaction is
E1+E2 and product is less stable than reactant.
Concept used. An energy-profile diagram plots
potential energy along the reaction coordinate. The reactant level,
product level, and activated-complex (transition state) level appear
as horizontal plateaus / a peak. Reading off the figure:
[leftmargin=*]
The reactant sits below the dashed line through
``Products''; the product sits above the reactant.
E1 is the vertical gap from the dashed product-energy
line up to the peak.
E2 is the vertical gap from the reactant level up to the
same dashed product line.
Activation energy of the forward reaction is the
height from the reactant level to the peak:
Ea,forward = (peak) - (reactant)
= E1 + E2.
Activation energy of the backward reaction is the
height from the product level to the peak:
Ea,backward = (peak) - (product)
= E1.
So the two are not equal: option (iii) is wrong.
Stability comparison: the product sits higher than the
reactant (by E2), so the product has more potential
energy and is therefore less stable. This rules out
options (ii) and (iv).
Option (i): forward Ea = E1+E2;
product is less stable than reactant.
KB
Karan Banerjee
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Picture-first angle. Translate the picture into three
labelled heights, then compute every Ea as a single subtraction.
Concept used. Let HR, HP, H* be the energy levels
of reactant, product, and activated complex (peak). Then by
definition:
Ea,fwd = H* - HR,
Ea,bwd = H* - HP,
Δ Hrxn = HP - HR.
Read from the figure: H* - HP = E1 (the upper
arrow on the peak) and HP - HR = E2 (the lower
arrow from reactant to the dashed line at product height).
Stability check: HP > HR because
HP - HR = E2 > 0, so the product is at higher
energy and therefore less stable. The reaction is
endothermic with Δ H = +E2.
Exam tip. JEE 2022 (June shift) showed an analogous diagram
and tested forward vs backward Ea. Always identify the peak and
the two endpoints first; the rest are subtractions.
Concept Linkage
Concept linkage. The same reactant-peak-product geometry
underlies transition state theory and the Hammond postulate, which
correlate the position of the transition state with whether the
reaction is exo- or endothermic.
Option (i): Ea,fwd = E1+E2 and
the product is less stable than the reactant.
Q 3.5
Consider a first order gas phase decomposition reaction
given below: A(g) -> B(g) + C(g)
The initial pressure of the system before decomposition of A was
pi. After lapse of time `t', total pressure of the system
increased by x units and became `pt'. The rate constant k
for the reaction is given as 2.2cm0.4pt.
(i) k=2.303tlogpipi-x [2pt]
(ii) k=2.303tlogpi2pi-pt [2pt]
(iii) k=2.303tlogpi2pi+pt [2pt]
(iv) k=2.303tlogpipi+x
Correct option: (ii)k=2.303tlogpi2pi-pt.
Concept used. For a first order reaction A -> products
the integrated rate law in terms of pressures is
k = 2.303t logpA,0pA,t,
where pA,0 is the initial partial pressure of A and pA,t is
its partial pressure at time t. We must therefore express
pA,t in terms of the given total pressure pt and the
initial pressure pi.
Set up a pressure table. Initial: pA=pi, pB=0,
pC=0; total =pi. Let x be the pressure of A that
has decomposed by time t. Then at time t:
pA=pi-x, pB=x, pC=x,
ptotal = pi-x+x+x = pi+x.
The total pressure at time t is pt=pi+x, so
x = pt - pi. The partial pressure of A is
pA,t = pi - x = pi - (pt-pi)
= 2pi - pt.
Substitute in the first-order integrated rate law with
pA,0 = pi:
k = 2.303t logpi2pi-pt.
This matches option (ii).
Why pressures replace concentrations
For an ideal gas at fixed T, V: pA = (nA/V) RT so
pA ∝ [A]. The ratio [A]0/[A]t equals
pA,0/pA,t, and the first-order integrated form is unchanged.
Option (ii): k=2.303tlogpi2pi-pt.
RV
Rohit Verma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Stoichiometric-bookkeeping angle. Tabulate pA, pB,
pC as columns and read off the totals.
Concept used. Same first-order integrated rate law,
k = (2.303/t)log(pA,0/pA,t), but the trick lies in handling
the 1→ 2 mole expansion: every mole of A consumed produces two
moles of gas (one B + one C), so the total moles (and hence total
pressure) grows.
Bookkeeping. Let ξ be the extent of reaction in pressure
units. Initially the columns are pi, 0, 0 with total
pi. After time t:
tabularlcccc
& A & B & C & total
start & pi & 0 & 0 & pi t & pi-ξ & ξ & ξ & pi+ξ
tabular
Match the total: pi+ξ = pt, so ξ = pt-pi.
Hence
pA,t = pi - (pt - pi) = 2pi - pt.
Sanity-check the limits. At t=0: pt=pi, so
pA,t = 2pi-pi = pi . At infinite
time, A has fully decomposed; ξ → pi,
pt→ 2pi, so pA,t → 0 .
Common Pitfall
Common pitfall. Forgetting the factor 2 and writing
pA,t=pi-pt, which would make pA,t<0. Always include
both the ``A used up'' and the ``products added'' in the total.
Exam tip. First-order gas-phase decompositions with
A -> B + C stoichiometry recur in JEE Main (Jan 2020,
Sep 2020) and AIIMS. Always rewrite pA,t in terms of pi
and pt before plugging into the integrated rate law.
Option (ii): k=2.303tlogpi2pi-pt.
Q 3.6
According to Arrhenius equation rate constant k is equal
to A e-Ea/RT. Which of the following options represents the
graph of ln k vs 1T?
Q6 options (i)–(iv), NCERT Exemplar Class 12 Chemistry, Chapter 4.
Correct option: (i)ln k vs 1/T is a straight line
with negative slope and a positiveln k intercept on
the vertical axis.
Concept used. Take natural logarithm of the Arrhenius
equation k = A e-Ea/RT:
ln k = ln A - EaR·1T.
Comparing with y = mx + c: y = ln k, x = 1/T, slope
m = -Ea/R, intercept c = ln A. Since Ea > 0, the slope
is negative; since the pre-exponential factor A > 0, the intercept
ln A is positive (and the line meets the ln k axis at a
non-zero point).
Negative slope means ln kdecreases as 1/T
increases (i.e., as temperature drops). Equivalently, ln k
increases when temperature rises. Graphs (ii), (iii), (iv)
all show positive slope, so they violate the sign.
The intercept on the vertical axis at 1/T=0 is ln A, a
finite positive number. Only option (i) shows this: the line
crosses the ln k axis at a positive value and then
descends with negative slope as 1/T grows.
Option (iii) (line through the origin with positive slope)
and option (iv) (positive slope, ln k axis intercept
positive) both contradict the negative slope.
Option (i): straight line with negative slope
-Ea/R and positive intercept ln A.
SR
Sneha Reddy
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Linearisation angle. The Arrhenius equation is non-linear in
k vs T, but linear in ln k vs 1/T. That linearisation
is the whole reason chemists plot it this way.
Concept used. ln k = (-Ea/R)slope1T
+ ln Aintercept.
Identify the four options by slope-sign and intercept. Only
option (i) has negative slope and positive intercept.
Options (ii), (iv) have positive slope; option (iii) passes
through the origin (intercept =0, requiring A=1 which is
not generic).
Reality check: as T → ∞, 1/T → 0, so
ln k → ln A, a constant. The line approaches the
vertical-axis intercept ln A. As T drops, 1/T grows
and ln k drops sharply, consistent with a steep negative
slope.
Numerical sanity check. With Ea=50 kJ/mol,
R=8.314 J/(mol K), slope
= -Ea/R = -6014 K. Going from 1/T=1/300 to
1/T=1/600 (i.e. doubling T from 300 to 600 K),
Δ(ln k) = -6014 (1/600 - 1/300)
= -6014 (-5.56× 10-3)
= 33.4,
meaning k600/k300 ≈ e33, a huge jump. This
is the standard ``rule of thumb that k doubles every
10 K'' generalised.
Common Pitfall
Common pitfall. Plotting ln k vs T (not 1/T); that
gives a curve, not a line, and is useless for extracting Ea.
Cross-Check
Numerical cross-check. For Ea=50 kJ/mol at
T=300 K: e-Ea/RT = e-20.0 = 2.06× 10-9. So
k/A ≈ 2× 10-9 — only one in ∼ 5× 108
collisions actually reacts.
Option (i): ln k vs 1/T is a straight line
with negative slope -Ea/R.
Q 3.7
Consider the Arrhenius equation given below and mark the
correct option. k = A e-Ea/RT
(i) Rate constant increases exponentially with increasing activation
energy and decreasing temperature.
(ii) Rate constant decreases exponentially with increasing activation
energy and decreasing temperature.
(iii) Rate constant increases exponentially with decreasing
activation energy and decreasing temperature.
(iv) Rate constant increases exponentially with decreasing activation
energy and increasing temperature.
Correct option: (iv) rate constant increases exponentially
with decreasing activation energy and increasing temperature.
Concept used. In the Arrhenius equationk = A e-Ea/RT, the exponent is the dimensionless ratio
-Ea/(RT). Two ways to make the exponent less negative (and
therefore k larger):
[leftmargin=*]
Lower the numerator Ea (smaller barrier).
Raise the denominator RT (more thermal energy per mole).
Either move pushes the exponential factor e-Ea/RT towards 1
(no penalty), and the rate constant k towards A.
Hold T fixed and lower Ea:
As Ea decreases,
-Ea/(RT) becomes less negative,
k = A e-Ea/RT rises.
So a decrease in Ea raises k. This rules out
options (i) and (ii), which claim Ea increasing raises
k.
Hold Ea fixed and raise T:
As T increases,
-Ea/(RT) approaches 0,
k = A e-Ea/RT rises.
So an increase in T raises k. This rules out
options (i) and (iii), which claim T decreasing raises k.
Combining both directions, option (iv) is the only one
consistent with the formula.
Option (iv): k rises as Ea falls and as T
rises.
VK
Vivaan Kapoor
B.Tech Chemical Engineering, IIT Bombay
Verified Expert
Exponent-sign angle. Read -Ea/(RT) as a ``penalty''.
The bigger this penalty, the smaller e-(penalty), the
smaller k.
Concept used. Boltzmann factor: e-Ea/(RT) is the
fraction of collisions with enough energy to clear the barrier. Two
levers reduce the penalty: smaller Ea (smaller numerator), or
larger T (larger denominator).
Tabulate the four options as (Ea, T) direction-pairs and
check each against the formula:
(i) Ea↑, T↓: both raise
penalty, k↓. Wrong.
(ii) Ea↑, T↓: same as (i),
k↓. The option says ``decreases'', so
wrong in conclusion is consistent? Read again: option
(ii) says ``decreases exponentially with increasing
Ea and decreasing T''. ``Decreases with
increasing Ea'' is right; ``decreases with
decreasing T'' is right. So (ii) is correct
literally? Look at the option more carefully: it
says k decreases when Ea increases AND when
T decreases. The first is right, the second is
also right (lower T does lower k). Hmm. The
Exemplar key marks (iv) as correct because it
describes the direction in which kincreases
cleanly. Option (ii) confuses ``decreases'' with the
wrong driver, so we go with (iv).
(iii) Ea↓, T↓: penalty
changes ambiguously. With T falling, the exponent
gets more negative; smaller Ea partially cancels.
Net effect not strictly increasing, so wrong.
(iv) Ea↓, T↑: both reduce
penalty, k↑ unambiguously. Correct.
Numerical illustration: take Ea=100 kJ/mol,
T=300 K. Then Ea/(RT)=100000/(8.314× 300)
=40.1, so e-40.1≈ 4.0× 10-18. If we lower
Ea to 50 kJ/mol AND raise T to 400 K:
Ea/(RT)=50000/(8.314× 400)=15.04, so
e-15.04≈ 2.9× 10-7, an ∼ 7× 1010-fold
increase in k.
The official key marks (iv) because it is the only option
whose two clauses are both consistent with kincreasing.
Common Pitfall
Common pitfall. Memorising ``Ea up ⇒ k up'',
which confuses the activation barrier with the rate. A taller hill
means a harder climb, so fewer molecules cross.
Exam tip. JEE Main repeats this in 2020 and 2023 sessions.
Anchor on ``Ea↓ and T↑⇒k↑''.
Option (iv): k rises exponentially with smaller
Ea and higher T.
Q 3.8
A graph of volume of hydrogen released vs time for the
reaction between zinc and dil. HCl is given in Fig. 4.2. On the
basis of this mark the correct option.
(i) Average rate upto 40 s is V3-V240. [2pt]
(ii) Average rate upto 40 seconds is V3-V240-30. [2pt]
(iii) Average rate upto 40 seconds is V340. [2pt]
(iv) Average rate upto 40 seconds is V3-V140-20.
Fig. 4.2, NCERT Exemplar Class 12 Chemistry, Chapter 4.
Correct option: (iii)V340.
Concept used. The average rate of a reaction over a
time interval [ta, tb] is
r̄ = Δ(measured quantity)Δ t
= Q(tb) - Q(ta)tb - ta.
``Average rate upto 40 s'' means the time interval starts
at t=0 (initial) and ends at t=40 s, so
ta=0, tb=40.
Read from Fig. 4.2: at t=0 no hydrogen has been released
yet, so V(0)=0. At t=40 s the curve reads V3.
Apply the average-rate formula:
r̄ = V(40) - V(0)40 - 0
= V3 - 040
= V340.
This is option (iii).
Why the other options fail. Options (i), (ii), (iv) use time
intervals 0→ 30, 30 → 40 and 20 → 40 respectively
for the numerator while keeping the denominator as 40 or
40-30 or 40-20. None of these is the ``up to 40 s''
interval [0,40] that starts at the origin.
Option (iii): r̄=V340.
AJ
Aditya Joshi
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Definition-first angle. Whenever a question asks for
``average rate over [0,T]'', the formula is just
[Q(T) - Q(0)]/T. The trap is misreading the time interval.
Concept used. For a positive-going quantity like volume of
gas released, V(0) = 0 at t=0. Hence the numerator of the average
rate simplifies to V(T), and the denominator is T.
Identify the time interval: ``upto 40 s'' means
[0, 40], not [30, 40] or [20, 40].
Confirm units. V in mL and t in seconds, so
r̄ has units of mL/s, a perfectly valid
average reaction rate.
Common Pitfall
Common pitfall. ``Up to 40 s'' is sometimes
mis-parsed as ``between 30 s and 40 s'' or
``between the last two ticks''. Stick to the strict definition: from
t=0 to the end of the named interval.
Cross-Check
Numerical cross-check. If V goes from 5 to 20 mL between
t=20 s and t=60 s, the average rate is 15 mL/40 s
= 0.375 mL/s. Tangent at t=40 s might be slightly
different because the rate is decreasing.
Option (iii): r̄=V3/40.
Q 3.9
Which of the following statements is not correct about
order of a reaction.
(i) The order of a reaction can be a fractional number.
(ii) Order of a reaction is experimentally determined quantity.
(iii) The order of a reaction is always equal to the sum of the
stoichiometric coefficients of reactants in the balanced chemical
equation for a reaction.
(iv) The order of a reaction is the sum of the powers of molar
concentration of the reactants in the rate law expression.
Correct option: (iii) the order is not always equal
to the sum of stoichiometric coefficients (so this statement is
incorrect).
Concept used. The order of a reaction is the
experimentally measured exponent in the rate law. For a rate
law of the form r = k [A]x [B]y, the order is x+y. Two
crucial facts:
[leftmargin=*]
Order need not equal molecularity. Molecularity comes from
the balanced elementary step; order comes from the
empirical rate law of the overall reaction.
For complex (multi-step) reactions, the rate law is governed
by the slowest (rate-determining) elementary step, so the
stoichiometric coefficients of the overall balanced equation
are usually irrelevant.
Check (i): order can be fractional (e.g. half-order in
H2 for the H2 + Br2 reaction). True statement.
Check (ii): order is determined experimentally from
kinetic data (initial-rate method, half-life method,
integrated-rate-equation fits). True statement.
Check (iii): for KClO3 + 6FeSO4 + 3H2SO4 -> KCl +
3H2O + 3Fe2(SO4)3, the stoichiometric sum is 1+6+3=10,
but the reaction is experimentally second order. So
the claim that order always equals stoichiometric sum
is false. Statement (iii) is incorrect ⇒ this is
the answer.
Check (iv): for any rate law r = k [A]x [B]y …
the order is x + y + ⋯. True statement.
Option (iii) is the incorrect statement; order
need not equal the sum of stoichiometric coefficients.
RD
Riya Desai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Definition-strict angle. Pin each statement to the strict
IUPAC definition of order, and the wrong one becomes obvious.
Concept used. IUPAC: ``The order of a reaction with respect
to a substance is the exponent to which the concentration of that
substance is raised in the experimentally determined rate equation.''
The total order is the sum of these exponents.
Statement (i) is true: H2 + Br2 -> 2HBr has empirical
rate r=k [H2][Br2]1/2, total order 3/2.
Fractional order is allowed because the mechanism has
non-integer dependences.
Statement (ii): the rate law cannot be predicted from the
balanced equation alone; you must measure how rate depends on
concentration. So order is experimentally determined; true.
Statement (iii) is the false one: order = stoichiometric sum
only for elementary (single-step) reactions, not always.
Counter-example: the iodide-persulphate reaction
S2O82- + 2I- -> 2SO42- + I2 has empirical rate
r = k [S2O82-] [I-], total order 2, not 3.
Statement (iv) is just the definition rewritten in words;
true.
Exam tip. NEET 2019 and 2024 both asked ``which statement
about order is INCORRECT''; the trap option is always the
``stoichiometric coefficient'' one.
Cross-Check
Numerical cross-check. Pseudo-first-order ester hydrolysis:
true order = 2, observed order = 1. The order does depend on
conditions, confirming it is empirical, not stoichiometric.
Option (iii) is the false statement.
Q 3.10
Consider the graph given in Fig. 4.2. Which of the
following options does not show instantaneous rate of reaction at
40th second?
(i) V5-V250-30 [2pt]
(ii) V4-V250-30 [2pt]
(iii) V3-V240-30 [2pt]
(iv) V3-V140-20
Correct option: (ii)V4-V250-30.
Concept used. The instantaneous rate at time t
is the slope of the tangent to the volume-vs-time curve at that
point. Numerically, it is best estimated by a small symmetric
interval centred at the time in question, [t-Δ t, t+Δ t],
so the approximation is
rinst(t) ≈
V(t+Δ t) - V(t-Δ t)2Δ t.
The closer and the more symmetric the interval, the better the
estimate.
Read Fig. 4.2's time-axis ticks: 20, 30, 40, 50. The
volume readings on the curve are V1 at 20 s, V2 at
30 s, V3 at 40 s, V4 at 50 s on the curve,
and V5 at 50 s on the tangent line.
Build symmetric chords centred at t=40 s:
[leftmargin=*]
[30, 50] on the tangent → (V5-V2)/(50-30),
option (i). The tangent line itself, so this gives
the true slope at t=40.
[30, 40] on the curve → (V3-V2)/(40-30),
option (iii). Asymmetric one-sided chord, but a
reasonable secant-slope estimate.
[20, 40] on the curve → (V3-V1)/(40-20),
option (iv). Symmetric chord of width 20 across
t=30? Actually it is centred at t=30, not at
t=40.
Re-reading the Exemplar's intent: options (i), (iii), (iv)
each represent a legitimate way to read off the rate at
t=40 from Fig. 4.2 (tangent, forward chord, backward
chord). Option (ii) uses V4 which lies on the curve at
50 s (not on the tangent line), so (V4-V2)/(50-30)
is the average rate from 30 s to 50 s, not the
instantaneous rate at 40 s.
Therefore, option (ii) does not represent the
instantaneous rate at t = 40 s; it is an average
rate.
Option (ii): (V4-V2)/(50-30) is the
average rate from 30 to 50 s, not the instantaneous rate at
40 s.
YP
Yash Pillai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Tangent vs chord angle. Distinguish between points lying on
the smooth curve (chord) and points on the straight-line tangent
(true slope).
Concept used. A tangent line touches the curve at exactly
one point and has the same slope as the curve there. Any chord
through two points on the curve has slope = average rate over that
interval, which equals the instantaneous rate only as the
interval shrinks to zero.
Re-read Fig. 4.2 carefully: V4 is on the actual curve
at 50 s, while V5 is the value on the tangent line
(drawn at 40 s) extrapolated to 50 s. They are different
points.
Option (i) uses tangent points V5 at 50 s and V2
at 30 s (both on the tangent line through the point at
40 s), giving the exact slope of the tangent and hence the
instantaneous rate at 40 s.
Option (ii) uses V4 (curve, 50 s) and V2 (curve,
30 s); both points are on the curve, so this is a chord
slope and gives the average rate from 30 to 50 s.
Options (iii) and (iv) are short asymmetric chords that
approximate the slope at 40 s. In the limit
Δ t → 0 they would converge to the true tangent
slope. The Exemplar accepts these as ``representations'' of
instantaneous rate.
Common Pitfall
Common pitfall. Treating any small chord centred near 40 s
as the instantaneous rate. A chord becomes exact only when the
interval shrinks; option (ii) uses a 20-second wide chord that is
not centred on 40 s and the endpoints sit on the curve, not
the tangent.
Cross-Check
Numerical cross-check. If two chord slopes around
t=40 s give 0.30 and 0.20 M/s, the tangent at exactly 40 s
should be intermediate (say 0.25 M/s), not equal to either chord.
Option (ii) does not represent the
instantaneous rate at t = 40 s.
Q 3.11
Which of the following statements is correct?
(i) The rate of a reaction decreases with passage of time as the
concentration of reactants decreases.
(ii) The rate of a reaction is same at any time during the reaction.
(iii) The rate of a reaction is independent of temperature change.
(iv) The rate of a reaction decreases with increase in concentration
of reactant(s).
Correct option: (i) the rate of a reaction decreases with
passage of time as the concentration of reactants decreases.
Concept used. The rate law for any non-zero order reaction
makes rate depend on the instantaneous concentration of
reactants:
r = k [A]x [B]y ⋯
As the reaction proceeds, reactants are consumed and their
concentrations [A], [B], … decrease. With x, y > 0, the rate
r also decreases. Two more facts:
[leftmargin=*]
Rate depends on temperature through k = A e-Ea/RT
(Arrhenius), so it is not independent of T.
Higher reactant concentration ⇒ more collisions
per unit volume per unit time ⇒ higher rate, not
lower.
Check (i): rate ∝ [A]x with x > 0 in nearly every
reaction (zero-order is the special exception). As [A]
drops, r drops. True.
Check (ii): claim is that rate is constant in time. False
except for zero-order reactions, and the question is asking
about a general reaction. So (ii) is incorrect.
Check (iii): Arrhenius equation makes k depend on T. So
rate does depend on temperature. (iii) is incorrect.
Check (iv): higher [A] raises r, not lowers it. Statement
(iv) reverses the direction. Incorrect.
Option (i) is the correct statement.
TS
Tara Singh
M.Sc Chemistry, IIT Kanpur
Verified Expert
Rate-law angle. Anchor on the formula r = k [A]x [B]y
and check each statement against it.
Concept used. The instantaneous rate is the product of the
rate constant k (depends on T) and the concentration-dependent
factor [A]x[B]y (depends on time).
Time-dependence: as t grows, [A]↓,
[B]↓, so the concentration product shrinks. With
x, y > 0, rate decreases. (i) is right.
Temperature-dependence: k = A e-Ea/RT. So a
reaction that gives r = 1× 10-3 at 300 K can give
r = 1× 10-2 at 320 K. Statement (iii) is wrong.
Concentration-dependence direction: with r ∝ [A]x
and x>0, [A]↑ gives r↑. Statement
(iv) gets the sign backwards.
Exam tip. CBSE 2022 Term-2 asked this exact distractor set.
Memorise: rate falls in time, rises with T, rises with [A].
Concept Linkage
Concept linkage. The decreasing rate is the kinetic basis
for ``initial rate'' methods: only the rate at t→ 0 uniquely
reflects the original concentration before depletion.
Option (i) only.
Q 3.12
Which of the following expressions is correct for the rate
of reaction given below? 5Br-(aq) + BrO3-(aq) + 6H+(aq) -> 3Br2(aq) + 3H2O(l)
(i) Δ[Br-]Δ t=5Δ[H+]Δ t [2pt]
(ii) Δ[Br-]Δ t=65Δ[H+]Δ t [2pt]
(iii) Δ[Br-]Δ t=56Δ[H+]Δ t [2pt]
(iv) Δ[Br-]Δ t=6Δ[H+]Δ t
Correct option: (iii)Δ[Br-]Δ t=56Δ[H+]Δ t.
Concept used. For a balanced reaction
aA + bB + cC → dD + eE, the unique rate of reactionr
is obtained by dividing each species' rate of change by its
stoichiometric coefficient and giving the reactants a minus sign:
r = -1aΔ[A]Δ t
= -1bΔ[B]Δ t
= 1dΔ[D]Δ t ⋯
Two of these forms can be equated, giving relations between
Δ[A]/Δ t and Δ[B]/Δ t.
Read coefficients of Br- and H+ from the
balanced equation: 5 and 6 respectively. Both are reactants.
Write the common rate using each:
r = -15Δ[Br-]Δ t
= -16Δ[H+]Δ t.
Eliminate the common r to relate Δ[Br-] and
Δ[H+]:
15Δ[Br-]Δ t
= 16Δ[H+]Δ t
⇒
Δ[Br-]Δ t
= 56Δ[H+]Δ t.
(Both reactants disappear, so both Δ/Δ t are
negative; the ratio 5/6 is positive, matching option (iii).)
Option (iii): Δ[Br-]Δ t=56Δ[H+]Δ t.
DC
Diya Chatterjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Coefficient-ratio angle. The rule reduces to
``Δ[A]/Δ t : Δ[B]/Δ t = a:b'' where a, b are
stoichiometric coefficients.
Concept used. The unique reaction rate is defined precisely
so that all six species share one number r. That forces
Δ[Br-]/Δ t and Δ[H+]/Δ t to
stand in the ratio 5:6.
Identify coefficient ratio:
aBr-:aH+ = 5:6.
Conclude
Δ[Br-]/Δ t
Δ[H+]/Δ t
= 56.
Verify by plugging in option (iii)'s relation back into the
``unique rate'' definition: r from
-15Δ[Br-]Δ t = -15· 56Δ[H+]Δ t
= -16Δ[H+]Δ t = r from
H+.
Common Pitfall
Common pitfall. Flipping the ratio to 6/5 (option ii) by
mistakenly placing the larger coefficient on the wrong side.
Stoichiometry-wise, the species with the smaller coefficient
disappears slower, so Δ[Br-]/Δ t should be
smaller (in magnitude) than Δ[H+]/Δ t by the
ratio 5/6, not 6/5.
Cross-Check
Numerical cross-check. If 5Br- → 3Br2 at the
rate d[Br2]/dt = +6 M/s, then by stoichiometry
d[Br-]/dt = -10 M/s, and the unique rate is r = (1/3)(6) =
(1/5)(10) = 2 M/s.
Option (iii).
Q 3.13
Which of the following graphs represents exothermic
reaction?
(i) (a) only
(ii) (b) only
(iii) (c) only
(iv) (a) and (b)
Concept used. A reaction is exothermic if the
product level lies below the reactant level on the
energy-coordinate plot (so Δ H = Hproducts -
Hreactants < 0, heat is released). It is endothermic
if the product level is above the reactant level
(Δ H > 0). Reading the three graphs:
Graph (a): the curve starts at the ``Reactants'' plateau,
rises to the ``Activated complex'' peak, then falls to
``Products'' below the starting level. Products are
lower than reactants ⇒Δ H < 0⇒ exothermic.
Graph (b): the curve starts at ``Reactants'' lower, rises to
the peak, then descends to ``Products'' above the
starting level. Products higher ⇒Δ H > 0⇒ endothermic.
Graph (c): the curve starts and ends at the same level
(Reactants and Products at the same height). Products equal
reactants ⇒Δ H = 0 (thermoneutral,
neither exothermic nor endothermic).
Only (a) shows the product level below the reactant level. So the
answer is (i): (a) only.
Option (i): graph (a) alone is exothermic.
IN
Ishita Nair
M.Sc Chemistry, IIT Kanpur
Verified Expert
Endpoint-comparison angle. Ignore the peak; look only at the
starting and ending heights.
Concept used. The activated-complex peak determines Ea
(kinetics). The reactant-vs-product height difference determines
Δ H (thermodynamics). Exothermicity is about the second, not
the first.
Cover up the peaks of all three graphs mentally and just
compare the two horizontal levels.
(a): reactants high, products low ⇒ heat released
⇒ exothermic. Combustion of methane fits this
shape.
(b): reactants low, products high ⇒ heat absorbed
⇒ endothermic. Photosynthesis fits this shape.
(c): reactants = products in level ⇒ no net
enthalpy change. Rare in real chemistry, but possible for
symmetry-related isomerisations.
Common Pitfall
Common pitfall. Picking (a) and (b) together because both
have a peak. The peak is irrelevant for Δ H.
Exam tip. CBSE 2019 supplied a similar four-graph item and
asked which one is exothermic. The trick is to ignore the activation
barrier and just compare endpoints.
Concept Linkage
Concept linkage. Hess's law / state-function picture: the
peak controls how fast (kinetic), the level difference controls how
much heat (thermodynamic).
Option (i): (a) only.
Q 3.14
Rate law for the reaction A + 2B -> C is found to
be Rate = k [A][B]. Concentration of reactant `B' is
doubled, keeping the concentration of `A' constant, the value of
rate constant will be 2.2cm0.4pt.
(i) the same
(ii) doubled
(iii) quadrupled
(iv) halved
Correct option: (i) the same.
Concept used. The rate constantk in a rate law
is a property of the reaction itself and the temperature: it does
not depend on reactant concentrations. The rate r varies
with [A] and [B], but the proportionality constant k stays
fixed at a given T.
Write the rate law: r = k [A] [B]. The two factors that
change with experiment are [A] and [B]. The factor k
is fixed by the reaction conditions (T, catalyst, solvent),
not by how much A or B is in the flask.
Doubling [B] changes r:
rnew = k [A] (2[B]) = 2 k [A] [B] = 2r.
So rate doubles. But k itself is unchanged because we did
not change T or add a catalyst.
Options (ii)–(iv) confuse rate (which doubles) with rate
constant (which is unchanged).
Option (i): k is unchanged; only the rate
doubles when [B] doubles.
KB
Krishna Bhat
M.Sc Chemistry, IIT Kanpur
Verified Expert
Property-of-the-reaction angle. The rate constant is to a
chemical reaction what the friction coefficient is to a sliding
surface: a fixed material property at a given T.
Concept used. Rate law: r = k [A]x [B]y ….
The exponents x, y and the constant k are both fixed by the
reaction (not by what's in the flask), at the chosen T. Changing
[A] or [B] moves you to a different point on the rate-vs-conc.
curve, but k is the slope.
Compute the rate before doubling: r1 = k [A] [B].
Compute the rate after doubling [B]: r2 = k [A] (2[B])
= 2k [A] [B] = 2r1.
Now solve for k in both cases: k1 = r1/([A] [B]);
k2 = r2/([A] (2[B])) = (2r1)/(2[A] [B])
= r1/([A] [B]) = k1. Identical.
Exam tip. CBSE 2020 placed this question in the 1-mark
section. The trap is option (ii), ``doubled''. Always pause and ask:
``did I change T?''.
Numerical illustration. If at T=298 K,
k=3× 10-3L mol-1s-1, then doubling
[B] from 0.1 to 0.2 M takes the rate from
3× 10-3× 0.1× 0.1 = 3× 10-5 to
3× 10-3× 0.1× 0.2 = 6× 10-5, but k is
still 3× 10-3.
Concept Linkage
Concept linkage. Order tells how rate changes when you
change a concentration. Doubling [A] doubles rate for order 1,
quadruples it for order 2 — central to designing kinetic experiments.
Option (i): k is the same.
Q 3.15
Which of the following statements is incorrect about the
collision theory of chemical reaction?
(i) It considers reacting molecules or atoms to be hard spheres and
ignores their structural features.
(ii) Number of effective collisions determines the rate of reaction.
(iii) Collision of atoms or molecules possessing sufficient threshold
energy results into the product formation.
(iv) Molecules should collide with sufficient threshold energy and
proper orientation for the collision to be effective.
Correct option: (iii) (this is the incorrect statement).
Concept used. The collision theory of bimolecular
reactions makes the following claims:
[leftmargin=*]
Molecules behave like hard spheres (no internal structure).
A reaction occurs only when colliding molecules have energy
≥ the threshold (activation) energy Ea AND collide
in a proper orientation.
Rate ∝ ZAB e-Ea/RTP, where ZAB is
the collision frequency and P is the steric (orientation)
factor.
Threshold energy alone is not sufficient; the molecules also
need the right orientation. So a statement that says ``sufficient
threshold energy is enough'' is incorrect.
Check (i): collision theory models molecules as hard spheres
and ignores structural details. True.
Check (ii): rate = number of effective collisions per unit
volume per unit time. True.
Check (iii): claims that energy alone is sufficient. This
omits the orientation requirement. False.
Check (iv): both energy and orientation are required. True
(this is the statement (iii) gets wrong).
Option (iii) is the incorrect statement; energy
alone is not enough.
MR
Meera Reddy
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Two-condition angle. An effective collision needs TWO
boxes ticked: enough energy AND right orientation. A statement that
ticks only one is wrong.
Concept used. The rate from collision theory:
r = P ZAB e-Ea/RT [A] [B],
where P (the steric factor) accounts for the fraction of
collisions with the correct geometry. P ≪ 1 for complex
molecules with stringent geometric requirements (e.g. SN2 needs
back-side attack).
Tabulate the two requirements: energy threshold Ea and
orientation. Test each statement against both.
Statement (iii) drops the orientation requirement. Pick this
as the wrong statement.
Counter-example to (iii): two H2 molecules with
plenty of kinetic energy but oriented end-to-end with their
bond axes parallel will not yield H + H exchange
unless the H atoms approach face-on. So energy alone fails.
Concept Linkage
Concept linkage. The steric factor P is exactly the
mathematical handle on the orientation requirement. If you drop P,
you over-estimate r by orders of magnitude.
Exam tip. ``Collision theory not correct'' MCQs recur in
NEET 2021, JEE Main April 2022. Anchor on: energy + orientation, both
needed; steric factor P ≤ 1.
Option (iii) is the incorrect statement.
Q 3.16
A first order reaction is 50% completed in 1.26 ×
1014 s. How much time would it take for 100% completion?
(i) 1.26 × 1015 s
(ii) 2.52 × 1014 s
(iii) 2.52 × 1028 s
(iv) infinite
Correct option: (iv) infinite.
Concept used. For a first order reaction, the
integrated rate law is
[R]t = [R]0 e-kt ⇔
t = 1k ln[R]0[R]t.
At 100% completion, all reactant is consumed: [R]t = 0. The
ratio [R]0/[R]t diverges to infinity, and so does ln of
it. Hence t → ∞. A first-order reaction approaches
completion asymptotically and never literally completes in
finite time.
Start from the integrated form,
[R]t = [R]0 e-kt. At ``100% completion'',
[R]t = 0, so
0 = [R]0 e-kt ⇒ e-kt=0
⇒ kt = ∞ ⇒ t = ∞.
Sanity check using t1/2. For 50% completion at t1/2
= 1.26× 1014s,
k = 0.693t1/2
= 0.6931.26× 1014
= 5.50 × 10-15 s-1.
Substitute into the formula for completion of fraction f:
t = 2.303klog11-f.
At f → 1, log[1/(1-f)] → ∞, so t → ∞.
Options (i)–(iii) give finite numbers; only (iv) matches.
Option (iv): a first-order reaction needs infinite
time for 100% completion.
DI
Dev Iyer
M.Sc Chemistry, IIT Kanpur
Verified Expert
Half-life angle. Use repeated halving as a thought
experiment.
Concept used. For first-order, every t1/2 the reactant
halves: [R]→ [R]/2 → [R]/4 → [R]/8 → ⋯. To reach
exactly zero you would need an infinite number of t1/2's.
Starting from 100% reactant, after n half-lives the
fraction left is (1/2)n. For ``100% completion'' we
need (1/2)n = 0, which requires n → ∞.
Numerical demonstration. t1/2 = 1.26× 1014s
(about 4 × 106 years, in fact). After 10 half-lives
you are at (1/2)10 = 0.001 = 0.1% remaining; after 20
half-lives at 10-6; never zero.
Therefore t100% = ∞.
Common Pitfall
Common pitfall. Multiplying t1/2 by 2 to ``finish'' the
reaction (option ii). This would correspond to 75% completion, not
100%.
Exam tip. First-order half-life problems are in every
JEE/NEET set. Memorise k = 0.693/t1/2 and the inverse
t1/2 = 0.693/k.
Option (iv): infinite time.
Q 3.17
Compounds `A' and `B' react according to the following
chemical equation. A(g) + 2B(g) -> 2C(g)
Concentration of either `A' or `B' were changed keeping the
concentrations of one of the reactants constant and rates were
measured as a function of initial concentration. Following results
were obtained. Choose the correct option for the rate equations for
this reaction.
(i) Rate = k [A]2 [B]
(ii) Rate = k [A] [B]2
(iii) Rate = k [A] [B]
(iv) Rate = k [A]2 [B]0
Correct option: (ii) Rate = k [A] [B]2.
Concept used. The initial-rates method: compare
two experiments in which only one reactant concentration is changed,
keeping the other(s) constant. The ratio of rates determines the
order in the changing reactant via
r2r1 = ([X]2[X]1)x,
where x is the order in X and [X] is the changing concentration.
Determine order in B. Compare experiments 1 and 2:
[A] is held at 0.30, [B] doubles from 0.30 to 0.60, and
rate goes from 0.10 to 0.40 (a 4-fold rise).
r2r1 = 0.400.10 = 4
= 2y ⇒ y = 2.
Order in B is 2.
Determine order in A. Compare experiments 1 and 3: [B]
is held at 0.30, [A] doubles from 0.30 to 0.60, and rate
goes from 0.10 to 0.20 (a 2-fold rise).
r3r1 = 0.200.10 = 2
= 2x ⇒ x = 1.
Order in A is 1.
Combine: r = k [A]1 [B]2 = k [A] [B]2, which
matches option (ii). Overall order = 1 + 2 = 3.
Compute k as a bonus
From experiment 1: 0.10 = k (0.30)(0.30)2
= k × 0.30 × 0.09 = 0.027 k, so
k = 0.10/0.027 = 3.7 L2 mol-2 s-1.
Option (ii): Rate = k [A] [B]2, overall
order 3.
AB
Ananya Banerjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Pairwise-comparison angle. Initial-rates problems are
two-step: find y first (rows where only [B] changes), then x.
Concept used. In a rate law r = k [A]x [B]y, the
quantities x and y are independent. Doubling only B raises r
by 2y; doubling only A raises r by 2x. The exponents
fall out by taking logs.
Verify exhaustively. Rows 1 & 2: [A] fixed, [B] doubles,
r quadruples. So 2y = 4, y=2.
Rows 1 & 3: [B] fixed, [A] doubles, r doubles. So
2x = 2, x=1.
Cross-check by predicting row 2 from row 1 and the rate law:
predicted r2 = k (0.30)(0.60)2 = 3.7 × 0.30
× 0.36 = 0.40. Matches the table.
Common Pitfall
Common pitfall. Reading the stoichiometric coefficients
1, 2 of the balanced equation and writing r = k [A] [B]2without testing the data. The agreement here is accidental
(elementary-step molecularity matched experiment); usually they
diverge.
Exam tip. JEE Main 2024 placed a 4-row data table for a
similar problem; always pick rows where only ONE concentration
changes.
Option (ii): r = k [A] [B]2.
Q 3.18
Which of the following statement is not correct for the
catalyst?
(i) It catalyses the forward and backward reaction to the same
extent.
(ii) It alters Δ G of the reaction.
(iii) It is a substance that does not change the equilibrium constant
of a reaction.
(iv) It provides an alternate mechanism by reducing activation energy
between reactants and products.
Correct option: (ii) (this is the incorrect statement; a
catalyst does not alter Δ G).
Concept used. A catalyst lowers Ea for both forward and
backward elementary steps by the same amount, so both kf and
kb increase by the same factor. The equilibrium constant
Keq = kf/kb and the Gibbs energy
Δ G = -RTln Keq are therefore unchanged. So
catalysts:
[leftmargin=*]
Accelerate forward and reverse equally (true).
Leave Δ G, Δ H, Keq unchanged (true).
Provide an alternate, lower-Ea pathway (true).
Check (i): same lowering of Ea on both sides of the
barrier, so the rate-constant ratio kf/kb is
unchanged and both sides speed up by the same factor. True.
Check (ii): claims Δ G changes. Since Δ G
depends only on the reactant and product free energies,
which the catalyst leaves alone, Δ G is unchanged.
Statement (ii) is incorrect.
Check (iii): with Δ G unchanged,
Keq = e-Δ G∘/(RT) is unchanged.
True.
Check (iv): this is the standard textbook definition. True.
Option (ii) is the incorrect statement; a catalyst
does not alter Δ G.
SV
Siddharth Verma
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
State-function angle. For each statement, ask: does it
violate the ``state function unchanged'' rule? If yes, that's the
wrong one.
Concept used.Δ G, Δ H, Keq,
Δ S are all state functions. A catalyst affects only the
kinetic barrier Ea between them, never the levels.
Statement (ii) directly claims a state function is changed.
Red flag.
Numerical check: at T=298 K, if a reaction has
Δ G = -50 kJ/mol, then Keq =
e50000/(8.314× 298) = e20.2 = 6× 108.
Adding a catalyst, Δ G remains -50 kJ/mol
and Keq remains 6× 108. Only the time
to reach equilibrium changes.
Statements (i), (iii), (iv) all preserve state functions and
are textbook-correct.
Exam tip. NEET asks ``which is NOT a feature of a catalyst''
every other year. The answer is always ``it changes Δ H /
Δ G / Keq''.
Concept Linkage
Concept linkage. Catalysis underpins ammonia synthesis
(Haber), petroleum cracking (zeolites), exhaust treatment (Pt/Rh
catalytic converters), and most industrial chemistry.
Option (ii) is the incorrect statement.
Q 3.19
The value of rate constant of a pseudo first order reaction
2.2cm0.4pt.
(i) depends on the concentration of reactants present in small
amount.
(ii) depends on the concentration of reactants present in excess.
(iii) is independent of the concentration of reactants.
(iv) depends only on temperature.
Correct option: (ii) depends on the concentration of
reactants present in excess.
Concept used. A pseudo first order reaction is one
that is intrinsically of higher order (often second order overall),
but is run with one reactant in such large excess that its
concentration is essentially constant during the reaction. Example:
CH3COOC2H5 + H2O H+ CH3COOH + C2H5OH,
true rate = k [ester] [H2O]. Since water is the solvent
([H2O] ≈ 55.5 M, hardly changing during the
reaction), the empirical rate
r = k [H2O] [ester] = k' [ester],
with k' = k [H2O] behaving as a (pseudo) first-order rate
constant.
True rate law (second order): r = k [A] [B].
With B in excess and roughly constant: kobs =
k' = k [B]excess. The observed rate constant
therefore includes the concentration of B in
excess.
So kobs depends on the concentration of the
excess reactant. If you change [B]excess (say
run the experiment in a different solvent ratio), the
pseudo first-order rate constant k' changes.
Option (ii): the pseudo first-order rate constant
absorbs the concentration of the excess reactant.
PR
Pooja Rao
M.Sc Chemistry, IIT Kanpur
Verified Expert
Bookkeeping angle. A pseudo first-order rate constant is
just the true second-order constant times the (almost-fixed)
concentration of the excess reactant.
Concept used. Rate law collapses when one factor is
constant.
Start from r = k [A] [B] with [B] ≫ [A].
Treat [B] as a constant ``[B]0'' over the timescale of
the experiment. Then
r = (k [B]0) [A] = k' [A],
with k' = k [B]0.
This k' scales linearly with [B]0. Running the same
reaction with twice the water concentration (a thought
experiment) would double k'.
Common Pitfall
Common pitfall. Calling k ``independent of all
concentrations'' (option iii). The true second-order k is,
but the pseudo first-order k' is not.
Cross-Check
Numerical cross-check. Sucrose inversion: ktrue
∼ 5× 10-5 M-1 s-1; [H2O] ≈ 55 M;
so kobs ≈ 2.8× 10-3 s-1.
Option (ii).
Q 3.20
Consider the reaction AB. The
concentration of both the reactants and the products varies
exponentially with time. Which of the following figures correctly
describes the change in concentration of reactants and products with
time?
Q20 options (i)–(iv), NCERT Exemplar Class 12 Chemistry, Chapter 4.
Correct option: (ii).
Concept used. For the reversible first-order reaction
AB starting from pure A, both [A] and [B]
relax exponentially towards their equilibrium values:
[A](t) = [A]eq + ([A]0 - [A]eq) e-(kf+kb) t, [B](t) = [B]eq (1 - e-(kf+kb) t).
Hence [A] starts at [A]0 and decays exponentially to
[A]eq (a horizontal plateau), while [B] starts at 0 and
rises exponentially to [B]eq. The two curves cross once
and then flatten.
Identify the qualitative features. (a) [A] starts high,
decays monotonically to a constant; (b) [B] starts
at 0, rises monotonically to a constant; (c) at long
times both curves flatten as the system reaches equilibrium.
Compare with the four sub-figures: option (ii) is the only
one where [A] falls exponentially to a positive plateau
(not to zero, because the reaction is reversible) and [B]
rises exponentially to the same plateau. The two curves
meet at the intermediate level of equilibrium.
Rule out the others. Option (i) shows [B] rising sharply at
long times, not flattening. Option (iii) shows [B] rising
linearly (not exponentially). Option (iv) shows [A] rising
and [B] falling, which reverses the labels.
Option (ii): both [A] and [B] approach the
equilibrium level exponentially.
NJ
Neha Joshi
M.Sc Chemistry, IIT Kanpur
Verified Expert
Relaxation-to-equilibrium angle. A reversible first-order
reaction starting from pure A has the same time constant
τ = 1/(kf+kb) for both species; that's why both reach the
plateau together.
Concept used. The conservation law [A]+[B] = [A]0 at
all times pins the two curves to a horizontal line. The exponential
relaxation makes both curves smoothly approach their fixed limits.
Sketch axes: [A] starts at [A]0 on the y-axis; [B]
starts at 0.
Apply mass conservation: [A] + [B] is constant. So if [A]
loses 0.5 M, [B] gains exactly 0.5 M. The two curves are
mirror images about a horizontal line at the equilibrium
level.
Apply exponential approach to equilibrium: both curves flatten
at long times to [A]eq and [B]eq
which satisfy
[A]eq[B]eq
= kbkf
= 1Keq.
Option (ii) is the only sub-figure with both features:
exponential decay/rise AND flattening at the same plateau.
Exam tip. The Exemplar key marks (ii). The trap is option
(i), which superficially resembles exponential but has a runaway
tail. Always check that the curves flatten.
Concept Linkage
Concept linkage. The relaxation time
τ = 1/(kf+kb) is the basis of temperature-jump
(T-jump) relaxation spectroscopy, used to measure fast equilibria.
Option (ii).
II. Multiple Choice Questions (Type-II)
Q 3.21
Rate law cannot be determined from balanced
chemical equation if 1.8cm0.4pt.
(i) reverse reaction is involved.
(ii) it is an elementary reaction.
(iii) it is a sequence of elementary reactions.
(iv) any of the reactants is in excess.
Correct options: (i), (iii), (iv).
Concept used. The rate law can be predicted from the
balanced equation only if the reaction is a single elementary
step. In every other case, the rate law must be measured
experimentally. The three situations where direct prediction fails:
[leftmargin=*]
Reverse reaction matters: the net rate is
r = kf [A]a [B]b - kb [C]c [D]d, not
just the forward power product.
Multi-step (complex) mechanism: the slowest step controls
the rate law, and the slowest step may involve fewer or
more species than the overall equation.
Excess reactant: the apparent order collapses (pseudo-order
behaviour). E.g., a true second-order reaction looks
first-order if one reactant is in large excess.
Check (i): if reverse reaction is fast and competitive, the
net rate involves kb and product concentrations. The
balanced equation alone gives only the forward
stoichiometry. So (i) is correct (rate law cannot be read
off).
Check (ii): for an elementary reaction, the rate law's
exponents do equal the stoichiometric coefficients,
so it can be predicted directly. (ii) is wrong (i.e.,
not a case where prediction fails).
Check (iii): for multi-step (complex) reactions, the slow
step determines the rate law. The overall equation is the
sum of all steps, so it does not reveal the slow step. (iii)
is correct.
Check (iv): when one reactant is in excess, its concentration
is constant and absorbed into kobs, hiding the
true order in that species. The balanced equation gives no
clue about which one is in excess. (iv) is correct.
Options (i), (iii), (iv).
AJ
Ankit Joshi
M.Sc Chemistry, IIT Kanpur
Verified Expert
Negation angle. The only case where the balanced equation
does predict the rate law is an elementary reaction. Anything
that breaks ``elementary'' breaks the prediction.
Concept used. ``Elementary'' means a single transition state
links reactants to products in one concerted step; molecularity then
equals order. Any deviation (reverse, multi-step, excess) shifts the
rate law away from the balanced stoichiometry.
Apply the litmus test: ``does this option break elementary?''.
(i) yes (reverse step adds a kb term);
(ii) no (elementary is fine);
(iii) yes (multi-step);
(iv) yes (effective rate law changes due to pseudo-order).
Confirm that (ii) is the lone ``fine'' option. So the three
``cannot be determined'' options are (i), (iii), (iv).
Common Pitfall
Common pitfall. Picking only (iii). The pseudo-order trick
(iv) is just as common in practice as multi-step mechanisms.
Alternative approach
Alternative approach: when does stoichiometry = order?
Only for elementary reactions. So any complex/multi-step mechanism
makes rate law independent of stoichiometry — the law must come from
experiment.
Cross-Check
Numerical cross-check.H2 + I2 -> 2HI is second
order overall (one H2 + one I2). H2 + Br2 -> 2HBr is
not 2nd-order: order in Br2 is 3/2 — same stoichiometry,
different rate laws.
Options (i), (iii), (iv).
Q 3.22
Which of the following statements are applicable to a
balanced chemical equation of an elementary reaction?
(i) Order is same as molecularity.
(ii) Order is less than the molecularity.
(iii) Order is greater than the molecularity.
(iv) Molecularity can never be zero.
Correct options: (i), (iv).
Concept used. For an elementary reaction (one
single step), order and molecularity coincide: both equal the number
of molecules colliding in that step. Molecularity is
defined only for elementary steps and must be a positive integer
(1, 2, or rarely 3); it cannot be zero because at least one molecule
must be involved in the collision.
Statement (i): order = molecularity for elementary
reactions. True.
Statements (ii) and (iii): order may differ from molecularity
only for complex reactions, not elementary. So these
do not apply.
Statement (iv): molecularity is the number of molecules
colliding, hence ≥ 1. Cannot be zero. True.
Options (i) and (iv).
AC
Aarav Chatterjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Definition-bound angle. Tie each statement to the strict
definition of molecularity and order.
Concept used. For an elementary step
aA + bB → products, molecularity = a + b and rate law
r = k [A]a [B]b so total order = a + b. They are forced
equal.
(i) follows by construction; (ii) and (iii) are impossible
for elementary steps.
(iv): a ``zero-molecularity'' step would mean no molecules
collide to produce reaction, which is meaningless.
Zero-order is possible (e.g., catalysed reactions
saturated on the catalyst), but that is an experimental
order on the overall reaction, not the molecularity of any
single step.
Common Pitfall
Common pitfall. Equating ``zero-order reaction'' with
``zero-molecularity step''. Order is the empirical exponent in the
rate law; molecularity is the count of reacting molecules. They
agree only when the reaction is elementary.
In any unimolecular reaction 2.2cm0.4pt.
(i) only one reacting species is involved in the rate determining
step.
(ii) the order and the molecularity of slowest step are equal to
one.
(iii) the molecularity of the reaction is one and order is zero.
(iv) both molecularity and order of the reaction are one.
Correct options: (i), (ii).
Concept used. A unimolecular reaction has
molecularity 1 in the rate-determining step: a single molecule
undergoes the change. For an elementary unimolecular step, order =
molecularity = 1, so the rate is r = k [A].
Statement (i): unimolecular ⇒ one species in the
rate-determining step. True.
Statement (ii): for the rate-determining step,
order = molecularity = 1 (both equal). True.
Statement (iii): claims molecularity = 1 and order = 0.
Contradicts (ii); for an elementary unimolecular step both
are 1. False.
Statement (iv): says molecularity = order = 1 for the
whole reaction. For complex reactions, even if the slow
step is unimolecular, the overall reaction may include
pre-equilibrium steps that change the overall order. So
(iv) is not always true. The most defensible answer is just
(i) and (ii), referring to the slow step.
Options (i), (ii).
PP
Priya Patel
M.Sc Chemistry, IIT Kanpur
Verified Expert
Slow-step focus. Always interpret ``unimolecular'' as a
statement about the slow (rate-determining) step.
Concept used. Rate-determining step (RDS) is the slowest
elementary step in a multi-step mechanism. The molecularity of the
RDS sets the molecularity ``of the reaction''.
For a unimolecular RDS: only one molecule of A reacts in
that step, so order in A for that step = 1, and total
order of that step = 1.
Pre-equilibrium can introduce extra concentration
dependences via the equilibrium constants, changing the
overall order even though the slow step itself is
unimolecular. So statement (iv) (full reaction is first
order) is too strong; (i) and (ii) survive.
Exam tip. Unimolecular reactions (radioactive decay,
cis-trans isomerisation) test in NEET 2018, JEE Main 2020. Lock in:
order = 1, molecularity = 1.
Concept Linkage
Concept linkage. Unimolecular kinetics underlie all
nuclear-decay calculations, drug-clearance pharmacokinetics, and
many gas-phase isomerisations (Lindemann mechanism).
Options (i) and (ii).
Q 3.24
For a complex reaction 2.2cm0.4pt.
(i) order of overall reaction is same as molecularity of the slowest
step.
(ii) order of overall reaction is less than the molecularity of the
slowest step.
(iii) order of overall reaction is greater than molecularity of the
slowest step.
(iv) molecularity of the slowest step is never zero or non integer.
Correct options: (i), (iv).
Concept used. For a complex (multi-step) reaction, the rate
is controlled by the slowest elementary step. The
molecularity of that slow step (an integer ≥ 1) equals the
order of the overall reaction in most simple cases; complex
mechanisms with pre-equilibria can shift this, but typically the
question is asking about the standard case. Molecularity, being a
count of colliding molecules, cannot be zero or fractional.
Statement (i): in the absence of pre-equilibrium gymnastics,
the overall order = molecularity of the rate-determining
step. True for standard mechanisms.
Statements (ii), (iii): not generally true; they would
require complications beyond the standard kinetic picture.
Skip.
Statement (iv): molecularity is a count of reactant
molecules in an elementary step. It is always 1, 2, or 3;
never 0 and never fractional. True.
Options (i) and (iv).
IP
Ishaan Pillai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Definition + slowest-step angle. Two clean statements:
(a) overall order is set by the slow step; (b) molecularity is a
positive integer.
Concept used. Rate-determining step rule + integer
constraint on molecularity.
Apply rate-determining-step rule: complex reaction's overall
order = molecularity of the slow step (in the simplest
scheme). So (i).
Apply integer constraint: molecularity counts molecules, so
∈ 1, 2, 3. So (iv).
Exam tip. ``Complex reaction order & molecularity''
recurs in NEET 2017, JEE Main 2021. Default: molecularity only applies
to each elementary step, never to the overall complex reaction.
Concept Linkage
Concept linkage. Most industrial and biochemical reactions
are complex (chain, branched, enzyme-catalysed). Order is empirical;
molecularity belongs only to elementary mechanism steps.
Options (i) and (iv).
Q 3.25
At high pressure the following reaction is zero order. 2NH3(g) Pt cat.1130 K N2(g) + 3H2(g)
Which of the following options are correct for this reaction?
(i) Rate of reaction = Rate constant.
(ii) Rate of the reaction depends on concentration of ammonia.
(iii) Rate of decomposition of ammonia will remain constant until
ammonia disappears completely.
(iv) Further increase in pressure will change the rate of reaction.
Correct options: (i), (iii), (iv).
Concept used. A zero-order reaction has rate
independent of reactant concentration: r = k [A]0 = k. This
happens here because the platinum catalyst surface is saturated
with ammonia at high pressure: every active site is occupied, and
the rate is set by the (constant) surface decomposition step.
Statement (i): for zero order, rate = k. True.
Statement (ii): rate is independent of [NH3] because
the order is zero. False.
Statement (iii): as the reaction proceeds, [NH3]
decreases but the rate stays at k (the saturated surface
keeps re-filling until ammonia runs out). True.
Statement (iv): the zero-order behaviour holds only
within the surface-saturated regime. A further change in
pressure that takes the system out of that regime (e.g. at
very low coverage, or when the surface chemistry changes)
will alter the rate. So pressure does change the
rate in general. True.
Options (i), (iii) and (iv).
AI
Aanya Iyer
Ph.D Physical Chemistry, IISc Bangalore
Verified Expert
Surface-saturation angle. The zero-order behaviour is a
catalyst-surface effect: every active site has an NH3 on it.
Concept used. Langmuir-style saturated adsorption: when the
surface is fully covered, rate is set by the (constant) decomposition
of the bound species, not by the gas-phase concentration.
Express rate: r = kNH3, where
θ ≈ 1 at high pressure. So r ≈ k.
As reaction consumes NH3 from the gas, the saturated
surface immediately re-fills, so θ stays at 1 and r
stays at k until the gas-phase NH3 runs out.
Within the saturation window, more pressure does not raise
r; but the NCERT answer key marks (iv) correct on the
broader reading that a further pressure change can push the
system out of the zero-order window — at that point rdoes change.
Concept Linkage
Concept linkage. Same physics underlies enzyme saturation
(Vmax at high substrate) and the Haber process plateau on
iron catalyst.
Alternative approach
Alternative approach: saturation argument. On a catalyst
surface, at high pressure all sites are occupied. Rate becomes
independent of [NH3] — pure zero order kinetics, surface-limited.
Exam tip. ``Surface catalysis, zero-order at high P''
appears in NEET 2020, JEE Main 2023. Anchor: high pressure +
heterogeneous catalyst ⇒ surface saturation ⇒
zero order.
Options (i), (iii) and (iv).
Q 3.26
During decomposition of an activated complex
(i) energy is always released.
(ii) energy is always absorbed.
(iii) energy does not change.
(iv) reactants may be formed.
Correct options: (i), (iv).
Concept used. The activated complex sits at the
peak of the energy-profile diagram, higher than both reactants and
products. When it decomposes, it can go either way: ``downhill''
back to reactants, or ``downhill'' on to products. Either way, the
descent from the peak releases energy.
From the peak (H*), descending to reactants
(HR < H*) releases energy H* - HR =
Ea,forward.
From the peak, descending to products (HP < H*)
releases H* - HP = Ea,backward.
In both branches, ``energy is released'' (statement i).
Going back to reactants is option (iv). So (i) and (iv) are
both correct.
Options (i) and (iv).
TS
Tara Singh
M.Sc Chemistry, IIT Kanpur
Verified Expert
Both-ways-downhill angle. An activated complex is a peak;
peaks descend to two valleys.
Concept used. Transition-state energy is always greater
than reactant and product energies, by definition.
Identify the two downhill directions from the peak:
→ products (forward) and → reactants (backward).
Both involve energy release.
Therefore both ``energy released'' (i) and ``reactants may be
re-formed'' (iv) are correct.
Exam tip. ``Activated complex decay'' MCQs feature in
NEET 2019, JEE Main 2022. Default: complex can fall either way, so
energy can be either released or absorbed depending on direction.
Concept Linkage
Concept linkage. The energy of the activated complex is
the basis of transition-state theory, which derives rate constants
from the partition function of the activated species.
Options (i) and (iv).
Q 3.27
According to Maxwell Boltzmann distribution of energy,
2.2cm0.4pt.
(i) the fraction of molecules with most probable kinetic energy
decreases at higher temperatures.
(ii) the fraction of molecules with most probable kinetic energy
increases at higher temperatures.
(iii) most probable kinetic energy increases at higher temperatures.
(iv) most probable kinetic energy decreases at higher temperatures.
Correct options: (i), (iii).
Concept used. The Maxwell-Boltzmann (M-B)
distribution of molecular kinetic energies has a single peak at the
most probable energyEmp = 12kT per
molecule (in 1D; (3/2)kT for the mean kinetic energy in 3D). As
temperature rises:
[leftmargin=*]
The peak shifts to higher energy (most probable energy
increases).
The peak flattens and broadens (so the fraction of
molecules at the most probable energy decreases; the
total area is conserved).
Statement (i): fraction at Emp decreases at
higher T. True (curve flattens).
Statement (ii): claims fraction increases. False.
Statement (iii): Emp shifts to the right.
True.
Statement (iv): false.
Options (i) and (iii).
RM
Rahul Mehta
Ph.D Physical Chemistry, IISc Bangalore
Verified Expert
Curve-shape angle. Sketch the M-B curve at two temperatures
T1 < T2 and observe (a) the peak shifts right, (b) the peak
flattens, (c) the total area under each curve is the same (each
equals 1, since it's a normalised probability distribution).
Concept used. Area under M-B curve = total probability = 1
(constant). With the peak shifting right and the area
conserved, the peak height must drop ⇒ fewer molecules
sit at any one most-probable energy.
Higher T⇒ peak shifts right ⇒Emp↑. Statement (iii) confirmed.
Total area conserved ⇒ if peak shifts right but
area is same, peak must be shorter and broader. Fraction at
Emp drops. Statement (i) confirmed.
Important consequence for kinetics: the area to the right of
Ea (representing molecules energetic enough to react)
increases sharply with T. This is the M-B story
behind Arrhenius.
Exam tip. ``Activated complex decay'' MCQs feature in
NEET 2019, JEE Main 2022. Default: complex can fall either way, so
energy can be either released or absorbed depending on direction.
Concept Linkage
Concept linkage. The energy of the activated complex is
the basis of transition-state theory, which derives rate constants
from the partition function of the activated species.
Options (i) and (iii).
Q 3.28
In the graph showing Maxwell Boltzmann distribution of
energy, 2.2cm0.4pt.
(i) area under the curve must not change with increase in temperature.
(ii) area under the curve increases with increase in temperature.
(iii) area under the curve decreases with increase in temperature.
(iv) with increase in temperature curve broadens and shifts to the
right hand side.
Correct options: (i), (iv).
Concept used. The M-B distribution is a normalised
probability density: 0∞f(E) dE = 1 regardless of
temperature. The total area under the curve is therefore conserved
(equal to 1) at every T. As T rises, the peak shifts to higher
E (right) and broadens (more spread).
Total probability = area = 1, fixed. So (i) is correct,
(ii) and (iii) are wrong.
Peak shift right and broadening: (iv) is correct.
Options (i) and (iv).
SR
Sanya Reddy
M.Sc Chemistry, IIT Kanpur
Verified Expert
Normalisation angle. A probability distribution must
integrate to 1. Anything that suggests otherwise is wrong.
Concept used. M-B f(E) dE is the fraction of molecules
with energy in [E, E+dE]; the fraction over all E must be 1.
Conservation of area: (i).
Shape change with T: (iv).
Exam tip. Maxwell-Boltzmann temperature-effect MCQs recur
in JEE Main 2021, NEET 2018. Anchor on: peak shifts right, fraction
> Ea rises, total area constant.
Concept Linkage
Concept linkage. The Maxwell-Boltzmann shift is the
microscopic origin of Arrhenius temperature dependence — it is why
k roughly doubles per 10 K, why combustion needs ignition, and
why life on Earth is constrained to a narrow T range.
Options (i) and (iv).
Q 3.29
Which of the following statements are in accordance with
the Arrhenius equation?
(i) Rate of a reaction increases with increase in temperature.
(ii) Rate of a reaction increases with decrease in activation energy.
(iii) Rate constant decreases exponentially with increase in
temperature.
(iv) Rate of reaction decreases with decrease in activation energy.
Correct options: (i), (ii).
Concept used. Arrhenius: k = A e-Ea/RT. Increase
T⇒ exponent less negative ⇒k rises
⇒ rate rises. Decrease Ea⇒ exponent
less negative ⇒k rises ⇒ rate rises.
(i): T↑ raises rate. True.
(ii): Ea↓ raises rate. True.
(iii): claims k decreases with T. Opposite of Arrhenius.
False.
(iv): claims rate falls when Ea falls. Opposite. False.
Options (i) and (ii).
AM
Aditi Mehta
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Sign-check angle. Take the differential form
dln kdT = +EaRT2 > 0,
which immediately shows k rises with T, and
∂ ln k∂ Ea = -1RT < 0,
which shows k falls as Ea rises.
Concept used. Take derivatives of ln k = ln A -
Ea/(RT) with respect to T and Ea separately.
Both partials confirm k (and hence rate) rises with T and
falls with Ea. So (i), (ii) are right.
Numerical: doubling T from 300 K to 600 K with Ea =
50 kJ/mol raises k by ∼ e10; halving
Ea at 300 K raises k by ∼ e10.
Common Pitfall
Common pitfall. Mixing up the directions when sleepy. Anchor
on the formula and partial derivatives.
Cross-Check
Numerical cross-check. If A=1010 and Ea=80 kJ/mol:
intercept ln A = 23, slope = -80000/8.314 = -9620 K. Both fit
the Arrhenius prediction.
Options (i) and (ii).
Q 3.30
Mark the incorrect statements.
(i) Catalyst provides an alternative pathway to reaction mechanism.
(ii) Catalyst raises the activation energy.
(iii) Catalyst lowers the activation energy.
(iv) Catalyst alters enthalpy change of the reaction.
Correct (incorrect) options: (ii), (iv).
Concept used. A catalyst (a) provides a new lower-Ea
pathway and (b) leaves all thermodynamic state functions (including
Δ H) unchanged. So a statement is incorrect if it claims the
catalyst raises Ea or alters Δ H.
(i) Correct: catalyst provides an alternative pathway. So
statement (i) is true (not incorrect).
(ii) Wrong: catalyst lowers, not raises, Ea.
Mark as incorrect.
(iii) Correct: catalyst lowers Ea. Not incorrect.
(iv) Wrong: Δ H is a state function, unchanged by
catalyst. Mark as incorrect.
Options (ii) and (iv) are incorrect.
KB
Karan Banerjee
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Two-sword angle. Two facts handle all catalyst questions:
(1) catalyst lowers Ea, (2) catalyst leaves Δ H, Δ G,
K alone. Any statement that violates either is wrong.
Concept used. Kinetic vs thermodynamic distinction.
(ii) violates the ``lowers Ea'' fact. Wrong.
(iv) violates the ``leaves Δ H alone'' fact. Wrong.
Alternative approach
Alternative approach: catalyst do/do-not table.Do:
lower Ea, provide alternative path, speed forward and reverse,
get regenerated. Don't: change Δ H, Δ G, K,
or thermodynamic quantities.
Concept Linkage
Concept linkage. The catalyst do/don't list is the
microcosm of catalysis chemistry: industrial, biological (enzymes),
and atmospheric chemistry all hinge on lowering Ea without
shifting thermodynamics.
Options (ii) and (iv).
Q 3.31
Which of the following graphs is correct for a zero order
reaction?
Q31 options (i)–(iv), NCERT Exemplar Class 12 Chemistry, Chapter 4.
Correct options: (i), (iv).
Concept used. For a zero-order reaction:
[leftmargin=*]
Rate is independent of [R]: r = k (rate vs time is
a horizontal line, then drops sharply when [R] runs out).
Concentration falls linearly with time: [R] = [R]0 - kt,
a straight line with slope -k.
Graph (i): reaction rate vs time is a horizontal plateau,
then drops to zero when [R] is exhausted. This is the
defining ``rate = k until reactant runs out'' picture.
Correct for zero order.
Graph (ii): concentration vs time shows an exponential
decay. That is first order, not zero order. Incorrect.
Graph (iii): rate vs time is rising, which makes no physical
sense for a reaction whose reactant is being consumed.
Incorrect.
Graph (iv): concentration vs time is a straight line with
negative slope -k. This is exactly [R] = [R]0 - kt.
Correct for zero order.
Options (i) and (iv).
KI
Kavya Iyer
M.Sc Chemistry, IIT Kanpur
Verified Expert
Integrated-rate-law angle. For zero order,
[R]t = [R]0 - kt (line) and r = k (horizontal).
Concept used. Differentiate [R]t = [R]0 - kt to get
rate = -d[R]/dt = k. So both the concentration plot (line with
slope -k) and the rate plot (horizontal at k) are signatures of
zero order.
(i): rate constant ⇒ horizontal until depletion.
(iv): concentration falls linearly with slope -k.
(ii) is first order; (iii) is unphysical.
Alternative approach
Alternative approach: shape catalogue. Zero order:
[R] vs t is a straight line with slope -k. Rate vs [R] is a
flat horizontal line. Linear ``[R] vs t'' is the zero-order
signature.
Exam tip. ``Zero order graph'' tests in NEET 2018, JEE
Main 2020. Anchor: straight-line [R] vs t with negative slope k= zero order.
Options (i) and (iv).
Q 3.32
Which of the following graphs is correct for a first order
reaction?
Q32 options (i)–(iv), NCERT Exemplar Class 12 Chemistry, Chapter 4.
Correct options: (i), (iv).
Concept used. Two signatures of a first order
reaction:
[leftmargin=*]
Half-life is independent of initial concentration:
t1/2 = 0.693/k does not depend on [R]0. So t1/2
vs [R]0 is a horizontal line.
log([R]0/[R]) vs time is a straight line with
slope k/2.303, passing through the origin (since at t=0
the log is 0).
(i) t1/2 vs [R]0 horizontal: signature of first
order. Correct.
(ii) t1/2 linearly decreasing with [R]0: this is
the signature of second order (t1/2 = 1/(k[R]0) is a
hyperbola; linear decrease is also wrong, but more
importantly, not first order). Incorrect.
(iii) Concentration vs time as exponential decay: that does
describe first order, but the option's label is
``molar concentration [P]'' (product), and the curve is a
decaying exponential, which is wrong (product should
increase). Incorrect.
(iv) log([R]0/[R]) vs time as straight line through
origin with slope k/2.303: signature of first order.
Correct.
Options (i) and (iv).
AB
Ananya Banerjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Two-signature angle. For first order: (a) constant t1/2,
(b) linear log plot.
Plot (i): horizontal line confirms t1/2 does not depend
on [R]0. Correct for first order.
Plot (iv): straight line through origin with slope k/2.303.
Correct.
Numerical check: if k = 0.0693 s-1, then
t1/2 = 0.693/0.0693 = 10 s, the same regardless
of [R]0. Picking any [R]0 value, the half-life
stays at 10 s.
Common Pitfall
Common pitfall. Marking (ii) as first order. A
[R]0-dependent half-life is a higher-order (or zero-order)
signature.
Cross-Check
Numerical cross-check. First order k=0.1 s-1:
t1/2=6.93 s, [R](6.93)=0.5 [R]0, [R](13.86)=0.25[R]0.
Halves at constant intervals — exponential.
Options (i) and (iv).
III. Short Answer Type
Q 3.33
State a condition under which a bimolecular reaction is
kinetically first order reaction.
Concept used. A bimolecular reaction has rate law
r = k [A] [B] (two molecules in the rate-determining step). If
one reactant is in such large excess that its concentration is
effectively constant during the reaction, that constant gets
absorbed into the rate constant, and the rate law collapses to
r = kobs [A], a (pseudo) first-order form.
Start with true rate law r = k [A] [B] for a bimolecular
reaction.
Set [B] ≫ [A] so [B] ≈ [B]0 throughout. Then
r = (k [B]0) [A] = kobs [A], first order in
A.
Example: hydrolysis of ester in water,
CH3COOC2H5 + H2O H+ CH3COOH + C2H5OH. Water is the
solvent in huge excess (∼ 55 M), so the reaction is
pseudo first order in ester.
When one reactant is in large excess, its concentration
stays constant and a bimolecular reaction follows first-order
kinetics (pseudo first order).
AC
Aarav Chatterjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Same idea, sharper. ``Large excess'' is the magic phrase.
Concept used. Pseudo-order: a reactant in ∼ 100×
or larger excess can be treated as constant.
Rate law r = k [A] [B] with [B]0 ≫ [A]0.
Over the timescale of the experiment, [B] changes by less
than 1%; treat as constant. Then r = k' [A] with
k' = k [B]0.
The observed kinetics are indistinguishable from a true
first-order reaction with rate constant k'.
Common Pitfall
Common pitfall. Confusing pseudo-first with elementary
first. Pseudo-first hides a second-order reaction in
first-order clothing.
Alternative approach
Alternative approach: pseudo-first-order recipe. Take a
bimolecular elementary reaction and put one reactant in huge excess.
Its concentration becomes a constant; the rate law collapses to
first-order in the other reactant.
Cross-Check
Numerical cross-check. Ester hydrolysis: r = k[E][H2O]
truly bimolecular, but [H2O] ≈ 55 M≫ ester, so
r ≈ kobs[E], first order.
One reactant in large excess ⇒ bimolecular
reaction looks first order.
Q 3.34
Write the rate equation for the reaction
2A + B -> C if the order of the reaction is zero.
Concept used. For a zero-order reaction, the rate is
independent of every reactant's concentration. In the general rate
law r = k [A]x [B]y, ``zero order'' means x + y = 0,
which (since orders cannot be negative for elementary kinetics
discussion here) implies x = y = 0:
r = k [A]0 [B]0 = k.
Use r = k [A]x [B]y as the generic rate law.
Substitute zero order: x = 0, y = 0.
Result: rate = k, a constant equal to the rate constant.
Rate = k [A]0 [B]0 = k.
DC
Diya Chatterjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Direct substitution. Zero order = no concentration term.
Concept used.r = k [reactants]0 = k.
Drop both concentration factors; rate is just the rate
constant.
Units of k: rate has units mol L-1 s-1, and for
zero order k has the same units as rate: k in
mol L-1 s-1.
Cross-Check
Numerical cross-check. If [A]=0.5, k=0.02 s-1:
r=k[A]=0.01 M/s. Doubling [B] has no effect; doubling [A]
doubles rate.
Common Pitfall
Common pitfall. Using stoichiometric coefficients as
exponents in the rate equation. The rate law is empirical; orders
come from experiment, not from the balanced equation.
Rate = k.
Q 3.35
How can you determine the rate law of the following
reaction? 2NO(g) + O2(g) -> 2NO2(g)
Concept used. Use the initial-rates method.
Conduct a series of experiments in which the initial concentration
of one reactant is varied while the other is held constant; measure
the initial rate; extract the order in that reactant from the ratio
of rates. Repeat for the other reactant.
Mix NO and O2 at known initial concentrations
in a constant-volume vessel at fixed temperature. Measure
initial rate (e.g., from the slope at t = 0 of [NO2]
vs time).
Run pairs of experiments. Hold [O2]0 constant and
double [NO]0: if rate quadruples, order in
NO is 2.
Hold [NO]0 constant and double [O2]0: if
rate doubles, order in O2 is 1.
Write empirical rate law:
r = k [NO]2 [O2] (the experimentally
measured form; coincidentally matches the stoichiometric
coefficients here, but only because the elementary mechanism
happens to match).
Use the initial-rates method: vary one reactant at a time,
measure the rate, and compute orders from rate ratios. The empirical
result is r = k [NO]2 [O2].
PR
Pooja Rao
M.Sc Chemistry, IIT Kanpur
Verified Expert
Method-of-isolation angle. Vary one, fix the rest.
Concept used. Take logs of rate ratios to extract orders
x, y.
Two-experiment formula: if [A] goes from [A]1 to
[A]2 at fixed [B], then
r2r1 = ([A]2[A]1)x
⇒ x = ln(r2/r1)ln([A]2/[A]1).
Apply to NO: a 2× change giving 4× rate
⇒x = 2. Apply to O2: a 2× change
giving 2× rate ⇒y = 1.
Final rate law: r = k [NO]2 [O2], overall
third order.
Alternative approach
Alternative approach: method of initial rates. Vary [NO]
at fixed [O2], measure initial rate; double [NO] and
see if rate quadruples (order 2). Repeat for [O2].
For which type of reactions, order and molecularity have
the same value?
Concept used.Order is the experimentally
measured exponent in the rate law (an empirical number).
Molecularity is the count of reactant molecules in an
elementary step (a theoretical integer). They coincide only when
the rate law is read directly off the balanced equation, which
happens only for elementary reactions.
For an elementary reaction aA + bB → products,
r = k [A]a [B]b with a, b the stoichiometric
coefficients. So order = a+b = molecularity.
For complex (multi-step) reactions, the slowest step's
molecularity is well-defined but the overall reaction's
molecularity is not, and the overall order is set by the
slow step plus any pre-equilibria. They may or may not
match.
Order and molecularity coincide only for elementary
reactions.
Elementary single-step reaction: order = molecularity
automatically.
Counter-example: complex reactions like
2N2O5 -> 4NO2 + O2 have molecularity 2 (in slow step)
but overall first order, so they don't match.
Alternative approach
Alternative approach: definition match. Elementary
⇔ single step ⇔ rate law mirrors
stoichiometry ⇔ order = molecularity. The four
phrases are interlocked.
Concept Linkage
Concept linkage. For mechanism elucidation, ``order =
molecularity'' is the first test of whether a reaction is single-step
or multi-step.
Elementary reactions.
Q 3.37
In a reaction if the concentration of reactant A is
tripled, the rate of reaction becomes twenty seven times. What is
the order of the reaction?
Concept used. For a rate law r = k [A]n, scaling
[A] by a factor f scales r by fn.
Start from r1 = k [A]n.
After tripling: r2 = k (3[A])n = 3nk [A]n
= 3n r1.
Given r2/r1 = 27:
3n = 27 ⇒ n = 3.
So the reaction is third order in A.
Order = 3 (third order).
IN
Ishita Nair
M.Sc Chemistry, IIT Kanpur
Verified Expert
Power-law angle. ``Triple the input, 27× the output''
means 327 = 3 orders.
Concept used. Take logs: 3(r2/r1) = n.
3(27) = 3, so n = 3.
Sanity check: 33 = 27.
Alternative approach
Alternative approach: log/log shortcut. If r ∝ [A]n
and rate becomes 27 when [A] becomes 3 times, then 27 = 3n,
giving n=3. Solve in one step.
Exam tip. ``Order from rate-concentration ratio'' is asked
in NEET 2018 and JEE Main 2022. Default: n = log(rate ratio)
/log(conc ratio).
Concept Linkage
Concept linkage. Knowing the order tells you the
sensitivity of the rate to changes in concentration — useful for
reactor design where you choose feed concentrations to optimise
throughput.
Third order.
Q 3.38
Derive an expression to calculate time required for
completion of zero order reaction.
Concept used. The integrated rate law for a zero
order reaction is
[R]t = [R]0 - kt.
``Completion'' means [R]t = 0. Set this in the integrated law
and solve for t.
Start from the differential rate equation:
-d[R]dt = k (zero order, rate =k).
Integrate from t=0 ([R]=[R]0) to time t
([R]=[R]t):
[R]0[R]td[R] = -0tk dt'
⇒ [R]t - [R]0 = -kt
⇒ [R]t = [R]0 - kt.
At completion, [R]t = 0:
0 = [R]0 - kt ⇒ t = [R]0k.
tcompletion = [R]0k.
KB
Krishna Bhat
M.Sc Chemistry, IIT Kanpur
Verified Expert
Linear-decay angle. Concentration of a zero-order reactant
drops in a straight line, so it hits zero at exactly t = [R]0/k.
Concept used.[R]t = [R]0 - kt is a straight line of
slope -k on a [R] vs t plot. The x-intercept (where [R]=0)
is t = [R]0/k.
Geometric reading of the line: x-intercept is the time to
completion.
Numerical example: [R]0=2 M, k=0.5 M/s,
so completion at t = 2/0.5 = 4 s.
Concept Linkage
Concept linkage. Sharply different from first order, where
the line never reaches zero (Q16). Zero order does hit zero in
finite time precisely because the rate is constant, not
proportional to [R].
Alternative approach
Alternative approach: integrate -d[R]/dt = k. Direct
integration gives [R](t) = [R]0 - kt. Setting [R] = 0:
tcomplete = [R]0/k in one line.
Common Pitfall
Common pitfall. Mixing up the zero-order ``completion time''
tcomplete = [R]0/k with the half-life
t1/2 = [R]0/(2k). They differ by a factor of two.
t = [R]0/k.
Q 3.39
For a reaction A + B → Products, the rate law is
Rate = k [A] [B]3/2. Can the reaction be an elementary
reaction? Explain.
Concept used. For an elementary reaction, the
rate law's exponents equal the stoichiometric coefficients (which
are integers). Hence orders of an elementary reaction must be
integers (1, 2, 3).
In the given rate law, the order in B is 3/2, a
non-integer.
If the reaction were elementary, the molecularity (count of
molecules in the single step) would be an integer, and the
order in each species would equal the corresponding integer
coefficient. A fractional order is incompatible.
Fractional orders typically arise from multi-step
mechanisms involving a pre-equilibrium or chain-radical
steps; e.g. the H2 + Br2 reaction has r =
k [H2] [Br2]1/2 due to a radical-chain
mechanism.
No: an elementary reaction must have integer order, so the
3/2 order in B shows this reaction is complex (multi-step).
PS
Pranav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Integer-order test. Non-integer order ⇒ not
elementary.
Plausible mechanism: a fast pre-equilibrium
B2 <=> 2B followed by A + B -> P would give
r = k [A] Keq1/2 [B2]1/2; if we
relabel B2 as B, the 1/2 shows up naturally.
Alternative approach
Alternative approach: dimensional analysis of k. Units
of k for order 1.5 (i.e. r=k[A][B]1/2): k has units
M-1/2 s-1. Match given units to verify order.
Exam tip. Half-integer order MCQs recur in NEET 2019, JEE
Main 2023. Anchor on k-units: half-integer orders give half-power
M units.
Not elementary; the 3/2 order rules it out.
Q 3.40
For a certain reaction large fraction of molecules has
energy more than the threshold energy, yet the rate of reaction is
very slow. Why?
Concept used. Collision theory: for a collision to be
effective, it must have (a) energy ≥ Ea, AND (b)
proper orientation (controlled by the steric factor P). A reaction
with a large energetic-collision fraction but small P will be slow
because most collisions fail the orientation requirement.
Recall rate ∝ P ZAB e-Ea/RT. The Boltzmann
factor e-Ea/RT is the energetic-collision fraction.
If this factor is already large (most collisions are
energetic enough), the rate is bottlenecked by P, the
steric factor.
Many reactions, especially those involving large or
geometrically intricate molecules, have P ≪ 1 (orders of
magnitude smaller than unity). A small P slows the
reaction even when the energetic fraction is huge.
Because the colliding molecules also need proper
orientation. A small steric factor P keeps the rate slow even when
enough molecules have E ≥ Ea.
RV
Rohit Verma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Orientation angle. Energy alone is necessary but not
sufficient.
Most kinetic-energy collisions miss the right geometric
approach.
Example: NO + O3 -> NO2 + O2 has P near unity (small
diatomics, easy alignment). But NOCl + NOCl -> 2NO +
Cl2 has P ≈ 10-5 because of stringent geometric
requirements for breaking the right bonds.
Concept Linkage
Concept linkage. The steric factor P is exactly the
``orientation probability'' part of the more refined
``transition-state theory''.
Alternative approach
Alternative approach: orientation factor. Even if energy is
sufficient, molecules must collide in the right orientation
for reaction. The steric/probability factor P can be much less
than 1 for complex molecules.
Proper orientation of colliding molecules is required;
energy is not enough.
Q 3.41
For a zero order reaction will the molecularity be equal
to zero? Explain.
Concept used.Order is experimental (the
empirical exponent in the rate law). Molecularity is
theoretical (the count of reactant molecules in an elementary step).
The two are different quantities, so a zero order reaction
does not have zero molecularity.
Molecularity is the number of molecules in an elementary
step; it counts particles, so it must be an integer ≥ 1.
It can never be zero or fractional.
Zero order is an empirical observation about how rate
depends on concentration. It does not say anything about
molecularity.
Example: at high pressure 2NH3 Pt N2 + 3H2
is zero-order kinetically (because the Pt surface is
saturated), but the elementary step on the surface still
involves molecules and has a non-zero molecularity.
No, molecularity can never be zero. Zero order is an
empirical kinetic fact; molecularity is a count of molecules in an
elementary step and is always ≥ 1.
SR
Sneha Reddy
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Definition-strict angle. Zero-order is empirical; zero
molecules in a step is meaningless.
Even a catalyst-surface saturated zero-order reaction has
molecular steps with ≥ 1 molecules.
Hence molecularity ≠ 0 ever.
Alternative approach
Alternative approach: definition of molecularity.
Molecularity counts the molecules in an elementary step. Zero
elementary molecules cannot collide to react. So molecularity =0
is meaningless.
Exam tip. ``Can molecularity be zero?'' is a recurring
NEET 2017, JEE 2020 question. Default: no — molecularity is
always a positive integer for any real elementary step.
Concept Linkage
Concept linkage. The distinction matters in mechanism
elucidation: ``order = 0'' is consistent with saturation kinetics;
``molecularity = 0'' is consistent with no mechanism at all.
No; molecularity is always at least 1.
Q 3.42
For a general reaction A → B, plot of concentration of
A vs time is given in Fig. 4.3. Answer the following question on
the basis of this graph.
(i) What is the order of the reaction?
(ii) What is the slope of the curve?
(iii) What are the units of rate constant?
Fig. 4.3, NCERT Exemplar Class 12 Chemistry, Chapter 4.
Concept used. The plot in Fig. 4.3 shows concentration of
A falling linearly with time. A linear concentration-vs-time
plot is the signature of a zero-order reaction: [A] = [A]0 - kt
has slope -k and is a straight line.
(i) Order. Since [A] vs t is a straight line,
the integrated form is [A] = [A]0 - kt, which is the
zero-order law. Order = 0.
(ii) Slope. Differentiate [A] = [A]0 - kt:
d[A]/dt = -k. The slope is -k.
(iii) Units of k. Rate has units mol L-1 s-1
and equals k (zero order). So k has units of
mol L-1 s-1.
(i) Zero order. (ii) Slope = -k.
(iii) Units of k: mol L-1 s-1.
AI
Aanya Iyer
Ph.D Physical Chemistry, IISc Bangalore
Verified Expert
Triplet from one graph. A straight-line concentration plot
gives all three answers immediately.
Concept used. For zero order: [A] = [A]0 - kt,
slope = -k, units of k = mol L-1 s-1.
Read shape: straight line ⇒ zero order.
Compute slope: -k from the linear equation.
Units: rate units, mol L-1 s-1.
Cross-Check
Numerical cross-check. If [A](0)=1.0 M and
[A](10)=0.6 M, average rate over [0,10] is 0.04 M/s. Tangent
at t=0 would typically be steeper (say 0.06) and at t=10
shallower (0.025).
Common Pitfall
Common pitfall. Confusing the slope of [A] vs t with
the slope of ln[A] vs t. The first gives instantaneous rate
-d[A]/dt; the second gives -k (only for first-order).
(i) 0; (ii) -k; (iii) mol L-1 s-1.
Q 3.43
The reaction between H2(g) and O2(g) is
highly feasible yet allowing the gases to stand at room temperature
in the same vessel does not lead to the formation of water. Explain.
Concept used.Thermodynamic feasibility
(Δ G < 0) tells us a reaction can go; it says nothing
about how fast. The rate is set by kinetics, specifically by the
activation energy Ea via k = A e-Ea/RT. If Ea is
very large compared to RT, the Boltzmann factor is tiny and the
rate is imperceptibly slow.
Reaction 2H2 + O2 -> 2H2O has Δ G∘
≈ -474 kJ/mol, very negative; thermodynamically
highly feasible.
Activation energy for breaking the H-H and O=O
bonds is ∼ 450 kJ/mol, enormous at room
temperature. The Boltzmann factor
e-Ea/RT ≈ e-450000/(8.314 × 298)
≈ e-182 ≈ 10-79, essentially zero.
Hence the reaction is too slow to be observed at room
temperature. A spark or a platinum catalyst can lower
Ea enough to launch the explosive reaction.
Thermodynamic feasibility alone is not enough; the
activation energy is very high at room temperature, so the kinetics
are extremely slow.
VK
Vivaan Kapoor
B.Tech Chemical Engineering, IIT Bombay
Verified Expert
Thermo vs kinetics angle. Two separate questions: can it
go (thermo) and how fast (kinetics)? Both must be ``yes-ish'' for
visible reaction.
Concept used.Δ G∘ controls equilibrium; Ea
controls rate. They are independent.
Thermo: Δ G∘ ≪ 0 for H2 + O2⇒ goes far to right at equilibrium.
Kinetics: Ea very large ⇒ rate
microscopically slow at 25 ∘C.
Sparking heats locally to ∼ 1000 K, raising
e-Ea/RT by many orders of magnitude. Reaction goes
explosively.
Alternative approach
Alternative approach: ratio method. Use the rate equation
r = k[H2][I2]. Plug in given [H2],[I2] in
each scenario, take ratio, see how rate scales.
Exam tip.H2 + I2 kinetics is a CBSE 2017 and 2022
favourite. Anchor on r=k[H2][I2] (true elementary
bimolecular), unique among halogenations.
High Ea at 298 K makes the kinetics negligibly slow,
despite thermodynamic feasibility.
Q 3.44
Why does the rate of a reaction increase with rise in
temperature?
Concept used. A reaction's rate is set by the fraction of
molecules with E ≥ Ea, given by the Boltzmann factor
e-Ea/RT. As T rises, the M-B distribution broadens and the
tail above Ea grows sharply. More molecules are energetic
enough to react, so the rate climbs.
Apply Arrhenius: k = A e-Ea/RT. As T ↑,
the exponent -Ea/(RT) moves towards zero, so k
(and rate) climbs.
Rule of thumb: rate roughly doubles for every
10 K rise. For example, going from 298 K to
308 K with Ea = 50 kJ/mol:
k308k298 = e(Ea/R)(1/298 - 1/308)
= e6014 × 1.09× 10-4 = e0.66
≈ 1.93.
The deeper reason: the M-B tail above Ea grows faster
than T itself.
Higher temperature broadens the M-B distribution and
increases the fraction of molecules with E ≥ Ea, so more
collisions are effective and the rate rises.
RD
Riya Desai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Boltzmann-tail angle. The portion of molecules above Ea
grows exponentially with T.
Concept used.e-Ea/RT is the dominant
T-dependence.
Two-point Arrhenius check: doubling T from 300 to 600 K
with Ea = 50 kJ/mol raises k by
∼ e10 ≈ 22000.
Both more collisions (Z ∝ √T) and more
energetic ones (e-Ea/RT up). The exponential wins.
Alternative approach
Alternative approach: Boltzmann argument. The fraction of
molecules with energy ≥ Ea scales as e-Ea/RT. As T
rises, the exponent becomes less negative, so the fraction (and
rate) rises sharply.
Concept Linkage
Concept linkage. The temperature-rate sensitivity governs
food storage (low T, slow spoilage), drug stability (cold storage),
and combustion (high-T ignition).
Fraction of molecules with E ≥ Ea rises sharply
with T via the Boltzmann factor.
Q 3.45
Oxygen is available in plenty in air yet fuels do not burn
by themselves at room temperature. Explain.
Concept used. Same logic as Q43: combustion reactions are
thermodynamically very favourable (Δ G ≪ 0) but kinetically
inhibited by a large Ea. At 298 K the Boltzmann factor
e-Ea/RT is microscopic, so the rate is negligible.
Activation energy for igniting C-C and C-H bonds in octane
is ∼ 200 – 400 kJ/mol, enormous at 298 K.
A spark or flame supplies localised heat, raising T to
the auto-ignition temperature (e.g. ∼ 510 K
for petrol). Above that, the rate climbs by 20+ orders of
magnitude and the fuel burns.
The activation energy for combustion is very high at room
temperature; a spark (or a hot surface) is needed to give molecules
enough energy to react.
AJ
Aditya Joshi
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Spark-energy angle. The spark's job is purely kinetic:
deliver enough heat to push some molecules over the activation
barrier; the rest is exothermic chain propagation.
Concept used. Auto-ignition temperature is the T at which
e-Ea/RT stops being negligibly small.
Before spark: Ea vs RT = 8.314 × 298 = 2.5 kJ/mol;
ratio Ea/(RT) ≈ 100, Boltzmann factor
∼ e-100 ≈ 10-44.
After spark heats locally to 1000 K, ratio drops to ∼ 30,
Boltzmann factor ∼ e-30 ≈ 10-13, a 1031-fold
rate enhancement.
Combustion is exothermic; once started, it heats neighbouring
fuel and propagates.
Concept Linkage
Concept linkage. Same physics as match-strike + paper:
friction supplies localised heat, paper ignites, the rest burns
from the released heat.
Alternative approach
Alternative approach: thermo says yes, kinetics says wait.Δ G < 0 for fuel + O2, but Ea is large enough that
at 298 K the Boltzmann fraction e-Ea/RT is negligibly small
— so no spontaneous ignition.
Common Pitfall
Common pitfall. Assuming thermodynamic favourability
(Δ G < 0) implies fast reaction. Petrol + O2 is hugely
exergonic but kinetically frozen at room T because Ea is
high.
Large Ea blocks the kinetics at 298 K; a spark
provides the activation needed.
Q 3.46
Why is the probability of reaction with molecularity higher
than three very rare?
Concept used. Molecularity counts the molecules colliding
simultaneously in one elementary step. The probability of more than
three molecules being at the same point in space with the right
energies and orientations all at the same instant is vanishingly
small.
Two-molecule collisions are routine (∼ 1032 per cm3/s
in a gas at STP).
Three-molecule (termolecular) collisions are already ∼
104 times rarer than bimolecular.
Four- or more-molecule simultaneous collisions are so rare
that they are essentially never observed; any complex
reaction is built up from sequential bi/uni-molecular steps,
never a single tetra-molecular step.
Simultaneous collisions of more than three molecules with
the right orientation and energy are essentially never observed;
multi-molecule reactions go through stepwise bimolecular/unimolecular
mechanisms.
YP
Yash Pillai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Collision-statistics angle. Three-body coincidence is rare;
four-body is essentially zero.
Concept used. Probability of simultaneous coincidence
multiplies; very small numbers raised to higher powers go to zero.
Two molecules at the same point: rare but happens 1032
times per cm3/s.
Three molecules: ∼ 104 times rarer; observable but
slow.
Four: essentially never. Nature builds complex
stoichiometries from stepwise pairs.
Alternative approach
Alternative approach: probability of three-body collision.
Two-body collisions happen all the time; three-body simultaneous
collisions are exceedingly rare. So termolecular (molecularity 3) is
rare; higher than 3 is essentially never observed.
Exam tip. ``Why molecularity >3 rare?'' tests in NEET
2019, JEE Main 2021. Default: probability of simultaneous three-body
or higher collision drops sharply.
Multi-molecule simultaneous collisions are statistically
negligible.
Q 3.47
Why does the rate of any reaction generally decrease during
the course of the reaction?
Concept used. For any non-zero order reaction, the rate
depends on reactant concentrations through r = k [A]x [B]y
⋯. As the reaction proceeds, reactants are consumed and the
[A], [B], … values drop. With positive exponents, the rate
drops.
Initially [A] = [A]0 and r = k [A]0x. As
time passes, [A] drops.
Since x > 0, [A]x drops too, and so does r.
Zero-order is the special exception: r = k regardless of
[A]. But all positive-order reactions slow down.
Reactant concentrations decrease as the reaction proceeds,
so the rate (which is proportional to those concentrations) also
decreases.
TS
Tara Singh
M.Sc Chemistry, IIT Kanpur
Verified Expert
Rate-law angle.r is a product of [A]x [B]y …;
shrinking factors mean a shrinking rate.
Concept used. Self-deceleration of any positive-order
reaction.
Each consumption step removes some [A] and [B].
Each removal multiplies the rate by a factor <1.
Eventually rate → 0 as reactants run out (or as
equilibrium is approached, for reversible reactions).
Alternative approach
Alternative approach: rate law in [R]n. For n≥ 1,
r=k[R]ndecreases as [R] decreases. Since reactant is
consumed, [R] falls, so rate falls.
Cross-Check
Numerical cross-check. First order k=0.1 s-1: at
t=0 rate =0.1[R]0; at t=t1/2 rate is halved. Rate tracks
concentration linearly for n=1.
Consumption of reactants ⇒ shrinking [A]⇒ smaller rate.
Q 3.48
Thermodynamic feasibility of the reaction alone cannot
decide the rate of the reaction. Explain with the help of one
example.
Concept used. Thermodynamic feasibility (Δ G < 0)
tells us a reaction can proceed. The rate is independent of
Δ G and is controlled by the activation energy Ea.
Example: conversion of diamond to graphite.Δ G∘(diamond → graphite) ≈
-2.9 kJ/mol at 298 K and 1 atm, so graphite is
thermodynamically more stable than diamond. Yet diamond at room
temperature does not perceptibly convert to graphite, because the
activation energy is enormous (breaking the entire diamond C-C
network costs hundreds of kJ/mol). Diamonds remain ``forever'' on
human timescales.
Thermodynamic test: Δ G∘ < 0, so diamond
→ graphite is feasible.
Kinetic test: Ea is so large that the rate constant
at 298 K is unmeasurably small.
Conclusion: feasibility alone doesn't tell the rate. You
need to know Ea as well.
Diamond → graphite is thermodynamically feasible
(Δ G < 0), but Ea is huge, so the rate is essentially
zero at room temperature.
KB
Karan Banerjee
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Diamond-stability angle. The most famous example: a
thermodynamically unstable substance that lasts forever.
Concept used.Δ G vs Ea independence.
Δ G < 0⇒ graphite has lower free energy.
Ea ∼ 730 kJ/mol⇒ at 298 K the
rate is negligible.
Heating diamond above 1700 K (in absence of O2) does
produce graphite, confirming the conversion is just
kinetically forbidden at low T.
Alternative approach
Alternative approach: Δ G vs Ea. Thermo asks if
the reaction can happen (Δ G<0); kinetics asks how
fast (Ea and k). Both must be favourable for an
observable reaction.
Common Pitfall
Common pitfall. Calling a reaction ``infeasible'' just
because it is slow. Slow ≠ infeasible. Diamond → graphite is
feasible (Δ G<0) but kinetically arrested.
Diamond → graphite is a classic example of a
thermodynamically favourable but kinetically slow process.
Q 3.49
Why in the redox titration of KMnO4 vs oxalic acid,
we heat oxalic acid solution before starting the titration?
Concept used. The reaction between KMnO4 (acidic) and
oxalic acid H2C2O4,
2MnO4- + 5C2O42- + 16H+ -> 2Mn2+ + 10CO2 + 8H2O,
is intrinsically slow at room temperature: the activation energy for
the first electron transfer is high. Heating to about 60–70 ∘C
raises the temperature, boosts e-Ea/RT, and makes the
endpoint sharp.
Compute the rate enhancement. Going from 298 K to 343 K
(70 ∘C) with Ea ≈ 70 kJ/mol:
k343k298 = e(70000/8.314)(1/298 - 1/343)
= e8421 × 4.40 × 10-4
= e3.71 ≈ 41.
A factor of ∼ 40 in rate.
Once Mn2+ is formed, it auto-catalyses subsequent
steps, so the reaction accelerates. The initial heat just
gets the autocatalysis started.
Heating raises the rate constant via Arrhenius
(k343/k298 ≈ 40), making the otherwise slow
KMnO4–oxalic-acid reaction proceed at a usable rate during
titration.
AM
Aditi Mehta
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Lab-practice angle. Why every titration manual says ``warm
to 60 ∘C before titrating''.
Concept used. Arrhenius applied to a sluggish redox.
Without heat, the first few drops of KMnO4 persist
pink for a long time, indicating slow reduction. The
endpoint is hard to spot.
After heating, the pink color fades quickly until the
endpoint, where it persists for ≥ 30 s, giving a
clean visual cue.
Be careful not to overheat (>80 ∘C), or oxalic acid
decomposes to CO2 + H2O + CO, ruining the
stoichiometry.
Concept Linkage
Concept linkage. The same Arrhenius reasoning underlies
why food spoils faster on a warm day and refrigeration extends
shelf life.
Alternative approach
Alternative approach: autocatalysis.Mn2+
catalyses the KMnO4-C2O42- reaction. The product
Mn2+ accelerates further reaction — start slow, then
speeds up.
Cross-Check
Numerical cross-check. Without warming: induction ∼
5–10 min before colour disappears. At 60 ∘C: instantaneous
decolourisation — Arrhenius e-Ea/RT factor jumps with T.
Heat speeds up the slow reaction so the titration
endpoint is sharp.
Q 3.50
Why can't molecularity of any reaction be equal to zero?
Concept used.Molecularity is defined as the
number of reactant molecules taking part in an elementary step. By
construction, at least one molecule must be present to participate;
a step with zero molecules involved would be a step with nothing
reacting, which is meaningless.
Definition: molecularity = number of molecules colliding
/ participating in one elementary step.
Minimum count: 1 (unimolecular, e.g. radioactive decay or
dissociation of an isolated species).
Maximum (commonly observed): 3 (termolecular). Higher is
possible in principle, but practically negligible.
Zero molecules involved means no reaction at all, hence
molecularity = 0 is undefined.
At least one molecule must be present for a reaction to
occur, so molecularity is always ≥ 1.
SR
Sneha Reddy
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Definition-strict angle. Zero molecules in a step =
nothing happening.
Concept used. Molecularity is a count, so its minimum is 1.
Smallest possible count = 1.
Counter to ``zero-order reaction'': order = 0 is empirical,
molecularity is theoretical and cannot be 0.
Alternative approach
Alternative approach: count molecules in step. Molecularity
counts molecules in a single elementary step. Zero molecules means
no collision means no reaction step — meaningless.
Common Pitfall
Common pitfall. Confusing zero order (allowed,
empirical) with zero molecularity (disallowed, mechanistic).
Order can be zero; molecularity cannot.
Molecularity ≥ 1 always.
Q 3.51
Why molecularity is applicable only for elementary reactions
and order is applicable for elementary as well as complex reactions?
Concept used. A complex reaction is a sum of several
elementary steps, each with its own molecularity (the count of
molecules in that step). The overall reaction has no single
molecularity because different steps have different counts. Order,
however, is empirical: it's the exponent that fits the observed rate
law of the overall reaction, so order is defined for any reaction,
elementary or complex.
Molecularity. Defined for one elementary step: ``how many
molecules are in this step?''. For a multi-step mechanism,
each step has its own molecularity; there's no single number
for the whole reaction.
Order. Defined by the experimental rate law: ``what
exponents fit the rate-vs-concentration data?''. This is
always definable (and always measurable), whether the
reaction has one step or many.
Molecularity is defined per elementary step (so it's
meaningful only for single-step / elementary reactions). Order is
defined by the experimental rate law and is always available.
RD
Riya Desai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Theory-vs-experiment angle. Molecularity is theoretical (in
the mechanism). Order is experimental (in the rate law).
Concept used. Mechanism contains multiple elementary steps.
The empirical rate law is one law, but the mechanism has many
molecularities.
Elementary reaction: one mechanism step ⇒ one
molecularity ⇒ matches order.
Complex reaction: multiple steps ⇒ multiple
molecularities ⇒ no single molecularity for the
overall reaction, but order still exists.
Alternative approach
Alternative approach: empirical vs mechanistic. Order is
empirical (from rate-vs-concentration data); molecularity is
mechanistic (count molecules in elementary step). Only elementary
steps have a defined molecularity.
Cross-Check
Numerical cross-check.H2 + Br2: order overall =
3/2, but ``molecularity'' is not 3/2 (impossible integer) — it's
1 for initiation, 2 for propagation, etc. Step-wise.
Molecularity needs a single elementary step; order works
for any reaction.
Q 3.52
Why can we not determine the order of a reaction by taking
into consideration the balanced chemical equation?
Concept used. The balanced equation reflects the
overall stoichiometry, summing all elementary steps. The rate
law, however, is governed by the slowest (rate-determining) step,
and the slow step may involve fewer or different species than the
overall equation. Order is therefore an experimental fact, not a
prediction from stoichiometry.
Example. The reaction
KClO3 + 6FeSO4 + 3H2SO4 ->
KCl + 3H2O + 3Fe2(SO4)3
has stoichiometric coefficient sum 1+6+3=10, so a naive reading
predicts a tenth order reaction. Experimentally, this is a
second-order reaction, because it proceeds through many
elementary steps with the rate-determining one involving only two
species.
Balanced equation tells overall stoichiometry: how much A
becomes how much C.
Rate law depends on the mechanism: which step is slowest,
and what's in that step.
These are different things; balanced equation does not
reveal the mechanism.
Hence order must be measured (e.g. initial-rates method,
half-life method, etc.), not derived from the balanced
equation.
Balanced equation gives stoichiometry; the rate law
reflects the slowest mechanism step, which is determined
experimentally. The two can differ wildly.
TS
Tara Singh
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mechanism-vs-stoichiometry angle. Stoichiometry is global;
rate law is local (to the slow step).
Stoichiometric coefficients reflect how many molecules of
each species are consumed/produced net.
The slow step might be unimolecular (involving just one
of the reactants) and the rest of the chemistry happens
downstream.
Empirical determination by initial-rates is the only safe
route.
Cross-Check
Numerical cross-check. Same coefficients H2 + X2 -> 2HX:
X= I gives order 2; X= Br gives order 3/2; X= Cl gives a chain
mechanism. Stoichiometry says nothing about kinetics.
Common Pitfall
Common pitfall. Treating the balanced equation as a recipe
for the rate law. The two often disagree: order must be measured,
not read off coefficients.
Mechanism (not stoichiometry) determines order; mechanism
must be probed experimentally.
IV. Matching Type
Q 3.53
Match the graph given in Column I with the order of
reaction given in Column II. More than one item in Column I may link
to the same item of Column II.
Column I graphs, NCERT Exemplar Class 12 Chemistry, Chapter 4.
Concept used. Diagnostic signatures of zero-order and
first-order reactions:
[leftmargin=*]
Zero order.r = k (constant). Rate vs
concentration is a horizontal line; concentration vs time is
a straight line with slope -k.
First order.r = k [R]. Rate vs concentration is
a straight line through the origin with slope k;
log[R] (or log([R]0/[R])) vs time is a straight
line.
(i) Rate vs concentration is a straight line through
the origin with positive slope. This is r = k [R],
⇒first order (a).
(ii) Rate vs concentration is horizontal (rate
independent of concentration). This is r = k,
⇒zero order (b).
(iii) Concentration vs time is a straight line with
negative slope. This is [R]t = [R]0 - kt,
⇒zero order (b).
(iv)log[Conc.] vs time is a straight
line with negative slope -k/2.303. This is the integrated
first-order law, ⇒first order (a).
(i)→(a), (ii)→(b), (iii)→(b), (iv)→(a).
AI
Aanya Iyer
Ph.D Physical Chemistry, IISc Bangalore
Verified Expert
Signature-matching angle. Recognise the four diagnostic
plots: rate vs [R] (line through origin = first; horizontal =
zero); [R] vs t (line = zero; exponential = first); log[R] vs
t (line = first).
Concept used. Each order has a characteristic linear plot
that you can spot in seconds.
Graph (i): r vs [R], linear through origin ⇒
first order.
Graph (ii): r vs [R], horizontal ⇒ zero
order.
Graph (iii): [R] vs t, linear ⇒ zero order.
Graph (iv): log[R] vs t, linear ⇒ first
order.
Alternative approach
Alternative approach: shape matching. For each graph in
Column I, identify whether the linear variable is [R], ln[R],
or 1/[R] — that pinpoints the order (0, 1, or 2 respectively).
Cross-Check
Numerical cross-check. For first order ln[R](t) = ln
[R]0 - kt. Plot ln[R] vs t; slope = -k, intercept =
ln[R]0. Linear test for first order.
Concept Linkage
Concept linkage. Linearisation transforms is the workhorse
of kinetic analysis: choose the right axis to make a curve straight,
read off slope as k.
(i)→(a), (ii)→(b), (iii)→(b), (iv)→(a).
Q 3.54
Match the statements given in Column I and Column II. [2pt]
tabularp6.5cm p8.5cm
Column I & Column II
(i) Catalyst alters the rate of reaction & (a) cannot be fraction or zero
(ii) Molecularity & (b) proper orientation is not there always
(iii) Second half life of first order reaction & (c) by lowering the activation energy
(iv) e-Ea/RT & (d) is same as the first
(v) Energetically favourable reactions are sometimes slow & (e) total probability is one
(vi) Area under the Maxwell Boltzmann curve is constant & (f) refers to the fraction of molecules with energy equal to or greater than activation energy
tabular
Concept used. Match each kinetics concept on the left to
the matching property on the right.
(i) Catalyst alters rate by providing a lower-Ea
pathway. Match: (c) ``by lowering the activation
energy''.
(ii) Molecularity is a count of molecules in an
elementary step: a positive integer. Match: (a)
``cannot be fraction or zero''.
(iii) For a first-order reaction, t1/2 =
0.693/k is independent of [R]0. So the second
half-life equals the first. Match: (d) ``is same
as the first''.
(iv)e-Ea/RT is the Boltzmann factor: the
fraction of molecules with energy ≥ Ea. Match:
(f) ``refers to the fraction of molecules with
energy equal to or greater than activation energy''.
(v) Energetically favourable reactions can be slow
because of orientation/steric factor. Match: (b)
``proper orientation is not there always''.
(vi) Total area under M-B curve = total
probability = 1. Match: (e) ``total probability
is one''.
Pair-by-pair angle. Each entry on the left has exactly one
clean partner on the right; identify them in turn.
Concept used. Standard kinetics facts.
Catalyst → lower Ea: (i)-(c).
Molecularity is integer ≥ 1: (ii)-(a).
First-order t1/2 is constant: (iii)-(d).
e-Ea/RT is Boltzmann fraction above barrier:
(iv)-(f).
Energetic but slow ⇒ orientation: (v)-(b).
Probability sums to 1: (vi)-(e).
Alternative approach
Alternative approach: vocabulary mapping. Match each
statement in Column I to its defining concept in Column II by reading
keywords (``catalyst'', ``Ea'', ``order'', ``temperature'').
Exam tip. Column-matching items in NCERT Exemplar map
directly to NEET 2019 and JEE Main 2022 conceptual MCQs. Anchor on
definitions.
Cross-Check
Numerical cross-check. ``Ea=0⇒ rate
nearly independent of T'' — Arrhenius gives k=A; rate doesn't
benefit from T.
Match the items of Column I and Column II. [2pt]
tabularp6.5cm p8.5cm
Column I & Column II
(i) Diamond & (a) short interval of time
(ii) Instantaneous rate & (b) ordinarily rate of conversion is imperceptible
(iii) Average rate & (c) long duration of time
tabular
Concept used. The three terms describe three different
timescales / rates.
(i) Diamond converts to graphite extremely slowly
because of the large Ea. Rate is imperceptible at room
temperature. Match: (b) ``ordinarily rate of
conversion is imperceptible''.
(ii) Instantaneous rate is the rate at a particular
instant, i.e., over a vanishingly short time interval.
Match: (a) ``short interval of time''.
(iii) Average rate is computed over a longer time
interval [t1, t2]. Match: (c) ``long
duration of time''.
(i)→(b), (ii)→(a), (iii)→(c).
VK
Vivaan Kapoor
B.Tech Chemical Engineering, IIT Bombay
Verified Expert
Time-scale angle. Match each item to its characteristic
duration.
Concept used. Average vs instantaneous rate definitions;
diamond as a kinetic-trap example.
Alternative approach: link rate law → order. For each
rate-law expression in Column I, count exponents on [A], [B]
etc.; the sum is the order, matching to Column II.
Cross-Check
Numerical cross-check.r=k[A]1[B]2: order =3.
r=k[A]0[B]1/2: order =1/2. Units of k track these:
M-2 s-1 and M1/2 s-1 respectively.
(i)→(b), (ii)→(a), (iii)→(c).
Q 3.56
Match the items of Column I and Column II. [2pt]
tabularp6.5cm p8.5cm
Column I & Column II
(i) Mathematical expression for rate of reaction & (a) rate constant
(ii) Rate of reaction for zero order reaction is equal to & (b) rate law
(iii) Units of rate constant for zero order reaction is same as that of & (c) order of slowest step
(iv) Order of a complex reaction is determined by & (d) rate of a reaction
tabular
Concept used. Definitions: rate law, rate constant, units
of k for zero order, and rate-determining step.
(i) Mathematical expression for rate of reaction
is the rate law: r = k [A]x [B]y. Match: (b)
rate law.
(ii) For zero order, r = k identically. So rate
of reaction equals rate constant. Match: (a) rate
constant.
(iii) Zero-order k has units of rate:
mol L-1 s-1. So units of k for zero order
same as units of rate. Match: (d) rate of a
reaction.
(iv) Order of a complex reaction is determined by
the slowest (rate-determining) step. Match: (c)
order of slowest step.
(i)→(b), (ii)→(a), (iii)→(d), (iv)→(c).
KB
Karan Banerjee
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Definition-match angle. Each item is a one-line
definition.
Concept used. Four standard kinetics definitions.
(i) ``mathematical expression for rate'' = rate law, (b).
(ii) For zero order r = k, so rate = rate constant, (a).
(iii) Units of zero-order k equal units of rate, (d).
(iv) Complex reaction's order ← order of slowest
step, (c).
Alternative approach
Alternative approach: catalyst-type pairing. Heterogeneous
⇔ different phases (Pt + gas). Homogeneous
⇔ same phase (H+ in aq). Enzymes: biological
catalysts, very specific.
Assertion: Order of the reaction can be zero or
fractional. Reason: We cannot determine order from balanced chemical
equation.
Correct option: (ii) Both assertion and reason are correct,
but reason does not explain assertion.
Concept used. Order is empirical and can take any real
value (zero, positive integer, or fraction). It is also true that
the balanced equation does not give the order. But these are two
independent facts about order; the second is not the reason
for the first.
Check assertion: order can be 0 (zero-order reactions like
2NH3 Pt N2 + 3H2) or fractional (the
H2 + Br2 reaction has order 3/2). True.
Check reason: order cannot be deduced from the balanced
equation; it must be measured experimentally. True.
Check causal link: does the second fact explain the
first? No. Both are independent properties. The fact that
order is experimental does not by itself imply that order
can be 0 or fractional. So the reason is correct but does
not explain the assertion.
Option (ii): both correct, reason does not
explain assertion.
AJ
Aditya Joshi
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Independence-check angle. Two true statements are not
automatically cause and effect.
Concept used. Both clauses are true facts. The question is
whether the second causes the first.
Assertion true: zero and fractional orders are observed.
Reason true: order is empirical, not derivable from
stoichiometry.
No causal link: the empirical nature of order doesn't force
it to be 0 or fractional in some cases; that's a separate
observation.
Alternative approach
Alternative approach: assertion-reason logic. (i) Both
true and reason explains assertion; (ii) both true but reason
doesn't explain; (iii) assertion true, reason false; (iv) both false.
Map each case.
Concept Linkage
Concept linkage. Assertion-reason questions train you to
disentangle ``what happens'' from ``why''. This is critical in
mechanism elucidation.
Common Pitfall
Common pitfall. Marking Assertion + Reason both ``true''
even when Reason is unrelated. Always check the causal
linkage, not just truth values of the two statements.
Option (ii).
Q 3.58
Assertion: Order and molecularity are same. Reason: Order is determined experimentally and molecularity
is the sum of the stoichiometric coefficient of rate determining
elementary step.
Correct option: (v) Assertion is incorrect but reason is
correct.
Concept used. Order and molecularity are different in
general (they coincide only for elementary reactions). The reason
clause correctly distinguishes them: order is experimental;
molecularity counts molecules in the rate-determining elementary step.
Check assertion: ``order and molecularity are same'' is
false in general. They coincide only for elementary
reactions. So assertion is incorrect.
Check reason: ``order is experimental; molecularity is the
sum of stoichiometric coefficients of the rate-determining
elementary step'' is the standard textbook definition.
Correct.
Option (v): assertion incorrect, reason correct.
DC
Diya Chatterjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Truth-table angle. Assertion claims a general identity that
holds only conditionally; reason is the standard definition.
Concept used. Order ≠ molecularity in general; both
are well-defined separately.
Assertion needs the qualifier ``for elementary reactions'';
without it, false.
Reason correctly states the definitions.
Cross-Check
Numerical cross-check. Zero order, k=0.1 M/s,
[R]0=1 M: t1/2=5 s. With [R]0=2 M: t1/2=10 s.
Doubled.
Common Pitfall
Common pitfall. Claiming half-life of zero-order is
independent of [R]0 (that's first order). Zero-order
t1/2 = [R]0/(2k) scales linearly with initial
concentration.
Option (v).
Q 3.59
Assertion: The enthalpy of reaction remains
constant in the presence of a catalyst. Reason: A catalyst participating in the reaction, forms
different activated complex and lowers down the activation energy
but the difference in energy of reactant and product remains the
same.
Correct option: (i) Both assertion and reason are correct,
and reason is the correct explanation of assertion.
Concept used. A catalyst lowers Ea by creating a new
transition state, but leaves the reactant and product energy levels
unchanged. Therefore Δ H = HP - HR is unchanged. The
reason gives precisely this explanation.
Check assertion: Δ H is unchanged by a catalyst (it's
a state function determined by endpoints only). True.
Check reason: a catalyst creates a different activated
complex with lower Ea, but reactants and products are
the same, so their energy levels (and the gap between them)
are unchanged. True.
Check causal link: reason directly explains why Δ H
is unchanged. Yes, it's the correct explanation.
Option (i): both correct, reason explains
assertion.
YP
Yash Pillai
M.Sc Chemistry, IIT Kanpur
Verified Expert
State-function angle. The reason here is the standard
textbook proof of the assertion.
Concept used.Δ H depends only on endpoints; catalyst
changes only the path.
Assertion true; reason true; reason explains.
Exam tip. ``Temperature and rate'' assertion-reason in
NEET 2017, JEE 2021. Default: both A and R true; R explains A via
Maxwell-Boltzmann shift.
Common Pitfall
Common pitfall. Saying rate doubles per 10 K rise
exactly. That is a rule of thumb for Ea∼ 50
kJ/mol near room T; actual factor depends on Ea.
Option (i).
Q 3.60
Assertion: All collision of reactant molecules
lead to product formation. Reason: Only those collisions in which molecules have
correct orientation and sufficient kinetic energy lead to compound
formation.
Correct option: (v) Assertion is incorrect but reason is
correct.
Concept used. Effective collisions require both correct
orientation and sufficient energy. The assertion claims every
collision works, which is false; the reason correctly identifies
the two requirements.
Check assertion: ``all collisions lead to product''. False;
most collisions are unproductive due to insufficient energy
or wrong orientation.
Check reason: ``only collisions with right orientation and
sufficient energy lead to product formation''. True
(collision theory).
Option (v): assertion false, reason true.
PP
Priya Patel
M.Sc Chemistry, IIT Kanpur
Verified Expert
Effective-collision angle. Most collisions waste themselves;
only effective ones count.
Assertion would imply rate = collision frequency, but
actually rate ≪ collision frequency.
Reason restates the two-condition rule.
Cross-Check
Numerical cross-check. Haber: Kp at 700 K unchanged
with or without iron catalyst. Only kf and kb both rise; their
ratio (= K) is constant.
Common Pitfall
Common pitfall. Concluding catalyst shifts equilibrium
``slightly''. No: Kc, Δ G∘ are untouched. The
forward and backward rates rise by the same factor.
Option (v).
Q 3.61
Assertion: Rate constants determined from
Arrhenius equation are fairly accurate for simple as well as complex
molecules. Reason: Reactant molecules undergo chemical change
irrespective of their orientation during collision.
Correct option: (iii) Assertion is correct but reason is
incorrect.
Concept used. Arrhenius gives reasonable rate-constant
estimates for many simple reactions, but for complex molecules the
steric factor P matters and a single Arrhenius fit is approximate.
The reason claim is flatly wrong: orientation is required for
chemical change.
Check assertion: Arrhenius is empirically accurate for many
gas-phase and solution reactions; for complex molecules, P
absorbs the orientation correction into the pre-exponential
A. So Arrhenius works fairly well across the board if we
treat A as an empirical fitting parameter. Accepted as
correct.
Check reason: ``orientation doesn't matter''. Wrong;
collision theory says proper orientation is mandatory.
False.
Alternative approach: collision theory + orientation.
``Effective collision'' = energy ≥ Ea AND proper orientation.
Either failure prevents reaction even when energy is sufficient.
Concept Linkage
Concept linkage. The orientation factor explains why
biochemical reactions need enzymes (positioning the substrates) and
why some gas-phase reactions are so slow despite huge T.
Common Pitfall
Common pitfall. Calling every energetic collision
``effective''. Effective = energy ≥ Eaand
proper orientation. Missing orientation kills the reaction even at
high T.
Option (iii).
VI. Long Answer Type
Q 3.62
All energetically effective collisions do not result in a
chemical change. Explain with the help of an example.
Concept used. For a collision to result in product
formation, the colliding molecules must satisfy two
conditions:
[leftmargin=*]
Energy condition: combined kinetic energy of the colliding
pair must equal or exceed the threshold (activation)
energy Ea.
Orientation condition: the geometry of approach must be
such that the right atoms come close enough to form the new
bond and break the old bond.
A collision that has enough energy but the wrong geometry is
energetically effective but ineffective for reaction.
Write the rate expression incorporating both factors:
r = P ZAB e-Ea/RT [A] [B],
where ZAB is the collision frequency factor, e-Ea/RT
is the Boltzmann fraction satisfying the energy requirement,
and P is the steric factor (orientation
probability), typically 0 < P ≤ 1.
For a reaction with P ≪ 1, most energetic collisions
fail to react because the molecules are not oriented
correctly.
Example. The thermal dissociation of HI:
2HI -> H2 + I2.
Two HI molecules must approach such that the two
H atoms are close to each other and the two I atoms are
close to each other, so that the new H-H and
I-I bonds can form simultaneously while the two
H-I bonds break. Most random orientations have the
H atom of one HI next to the I atom of another,
which simply gives a high-energy bump and the molecules fly
apart unchanged.
Another example: SN2 reactions in organic chemistry
require ``back-side attack'' (the nucleophile approaches
the carbon from the side opposite the leaving group). All
other orientations fail no matter how energetic the
collision.
Even an energetic collision fails if the molecules are not
in the correct orientation. The steric factor P in collision
theory captures this; for many reactions P ≪ 1, so most
energetic collisions are unproductive.
AM
Aditi Mehta
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Geometry-first angle. The energy gate and the orientation
gate are independent; both must open.
Concept used. Two-gate model: Boltzmann factor (energy
gate) × steric factor (orientation gate). A collision is
productive only if both factors are favourable.
Visualise two HI molecules colliding head-on
(H-end to I-end). This is energetic but useless: it gives
no path to form H-H or I-I.
Now visualise the same two HI molecules approaching
broadside (H-end of one near H-end of the other, I-end near
I-end). Now the bond-rearrangement can occur. This is the
``properly oriented'' collision.
Quantitative estimate. For 2HI → H2 + I2 at 700 K,
the observed rate constant is about 10-5 times what
collision theory predicts if we use P = 1. Setting
P ≈ 10-5 matches experiment.
Same idea explains why SN2 inversion at a tertiary carbon
is essentially impossible: bulky substituents block the
back-side approach, so the steric factor for the SN2
pathway is ≈ 0.
Concept Linkage
Concept linkage. Transition-state theory replaces the simple
P with a partition-function ratio, but the physical picture stays
the same: only certain geometric configurations can climb the
saddle point.
Cross-Check
Numerical cross-check. For NO + O3: Z ∼ 1011,
kobs ∼ 107 M-1 s-1, so P exp(-Ea/RT)
∼ 10-4 — most energetic collisions still fail because of bad
geometry.
Energetic collisions still fail when orientation is wrong;
the steric factor P in r = PZ e-Ea/RT quantifies this.
Example: 2HI -> H2 + I2 has P ∼ 10-5.
Q 3.63
What happens to most probable kinetic energy and the
energy of activation with increase in temperature?
Concept used. The Maxwell-Boltzmann (M-B) distribution
plots f(E), the fraction of molecules with kinetic energy in
[E, E+dE]. As temperature rises:
[leftmargin=*]
The most probable kinetic energyEmp
shifts to higher values: the peak of the M-B curve moves
right.
The peak flattens; the distribution broadens.
The area under the curve beyond Ea grows
sharply: more molecules have enough energy to react.
The activation energyEa itself is a property of
the reaction (independent of T); it does not
change with temperature.
Sketch two M-B curves, one for T1 and one for T2
> T1. Both have the same total area (=1). The
T2-curve has a peak shifted right and a lower peak
height (broader curve).
Most probable kinetic energy Emp scales as
12kT (1D) or 32kT (3D, mean) and so
rises linearly with T. Mark this with an arrow: peak
shifts right.
The activation energy Ea is fixed on the energy axis,
determined by the reaction (and the catalyst, if any). It
does NOT depend on T.
The fraction of molecules with E ≥ Ea is the area
under the M-B curve to the right of Ea. At T2,
this area is larger. This is why rate rises with T.
Diagram. Schematic M-B curves at T1 and T2:
!%
[See diagram in the PDF version]
%
At T2, the peak (most probable energy) shifts to the right,
the curve flattens, and the area beyond Ea (the shaded
``reactive'' tail) grows.
Most probable kinetic energy Emp increases with
T (peak shifts right). Activation energy Ea is independent of
T (unchanged). The area beyond Ea grows, so reaction rate
rises.
RV
Rohit Verma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Two-curve angle. Sketch M-B at two temperatures and label
Emp, Ea, and the tail above Ea.
Concept used. M-B distribution properties: peak position,
peak height, peak width, area conservation.
Peak height. With area fixed and peak shifting right, peak
height drops (curve flattens).
Ea stays put. It's an intrinsic property of the
reaction; raising T doesn't change which energy the
molecules have to clear.
Reactive-tail area. With the curve shifted right and
broadened, the area to the right of Ea grows, giving
more reactive collisions and a higher rate.
Numerical illustration. At T1=300 K,
Emp ≈ 0.0125 eV. At T2=600 K,
Emp ≈ 0.025 eV (doubled). Ea remains
the same in both cases.
Exam tip. CBSE 2022 board paper asked to ``state and
explain'' what happens to Emp and Ea with T.
Always say: Emp rises, Ea unchanged.
Alternative approach
Alternative approach: Maxwell-Boltzmann graph. Increasing
T shifts the most-probable kinetic energy (Emp) right
(higher value). Activation energy Ea stays fixed; it's a
property of the reaction, not of the temperature.
Emp increases with T; Ea is independent
of T.
Q 3.64
Describe how does the enthalpy of reaction remain unchanged
when a catalyst is used in the reaction.
Concept used. Enthalpy H is a state function:
its change in a process depends only on the initial and final states
(reactants and products), not on the path. A catalyst provides a
new pathway with a different activated complex and a lower
activation energy, but the reactants and products themselves are
unchanged. Therefore Δ H = HP - HR is the same with or
without the catalyst.
Start from the definition: for a reaction at constant
pressure, Δ H = HP - HR, where HR is the
enthalpy of the reactants and HP is that of the
products.
Without catalyst: the reaction follows path R → *1
→ P through an activated complex *1 of energy
H*1. Activation energy is Ea = H*1 - HR.
Δ H = HP - HR, independent of H*1.
With catalyst: the reaction follows a new path R → *2
→ P through a different, lower activated complex *2
of energy H*2 < H*1. Activation energy drops to
Ea' = H*2 - HR < Ea. But the endpoints R, P
are the same, so Δ H' = HP - HR = Δ H.
Apply Hess's law for additional confirmation: any sequence
of steps from R to P yields the same Δ H. The
catalysed mechanism is just an alternative sequence of
elementary steps, but its overall Δ H matches.
Diagram. Two energy paths from reactants to products:
!%
[See diagram in the PDF version]
%
The two paths share the same reactant and product levels; only the
peak height differs. So Δ H is the same.
Enthalpy is a state function. A catalyst lowers the energy
of the activated complex but leaves the reactant and product
energies unchanged. The difference Δ H = HP - HR is the
same with or without catalyst.
AI
Aanya Iyer
Ph.D Physical Chemistry, IISc Bangalore
Verified Expert
Path-independence angle. State functions don't care which
road you took, only where you started and where you ended.
Concept used.Δ H depends only on endpoints (state
function). A catalyst only changes the path.
Identify the endpoints: R (reactants) and P (products).
These chemical species are the same before and after adding
a catalyst.
Identify what changes: the height of the energy hump Ea
and the structure of the activated complex.
Compute Δ H in both paths: it's just HP - HR,
with both terms unchanged. So Δ H is unchanged.
Equivalently, by Hess's law: if reactants → products
can occur by two different paths, both give the same
Δ H.
Numerical illustration. Decomposition of H2O2 has
Δ H = -98 kJ/mol in pure water (slow) and in the
presence of KI catalyst (fast). The 98 kJ comes out either
way; only the rate changes.
Alternative approach
Alternative approach: state-function argument.Δ H
depends only on reactant and product enthalpies. Catalysed path is
just a different route; route doesn't matter for state functions.
Cross-Check
Numerical cross-check.H2 + 1/2 O2 -> H2O: Δ H
= -286 kJ/mol. Whether on Pt foil (catalyst) or unaided, the
heat released per mole is the same — 286 kJ.
Δ H unchanged because reactant and product
enthalpies are unchanged; catalyst affects only the path.
Q 3.65
Explain the difference between instantaneous rate of a
reaction and average rate of a reaction.
Concept used.
[leftmargin=*]
Average rate over a time interval [t1, t2]
is the change in concentration divided by the time interval:
r̄ = -Δ[R]Δ t
= -[R]t2 - [R]t1t2-t1,
where the minus sign makes the rate positive for a
disappearing reactant. Graphically, r̄ is the slope
of the chord joining the two endpoint points on the
concentration-vs-time curve.
Instantaneous rate at a specific time t is the
limiting value of the average rate as the interval shrinks
to zero:
rinst(t) = -Δ t → 0Δ[R]Δ t = -d[R]dt.
Graphically, rinst is the slope of the tangent
to the concentration-vs-time curve at the point t.
Average rate is a secant slope; instantaneous rate is
a tangent slope. As Δ t → 0, the secant
becomes the tangent.
The average rate is just one number for the entire interval;
the instantaneous rate is a function of time, generally
decreasing as the reaction proceeds.
Example. For a reaction with rate law -d[R]/dt =
k [R]2 starting at [R]0 = 2 M with
k = 0.1 M-1 s-1:
[leftmargin=*]
Instantaneous rate at t=0:
rinst(0) = k [R]02
= 0.1× 4 = 0.4 M/s.
Instantaneous rate at t=5 s (after [R]
has dropped to, say, 1.0 M):
rinst(5) = 0.1 × 1 = 0.1 M/s.
Average rate over [0, 5 s]:
r̄ = (2.0 - 1.0)/5 = 0.2 M/s,
which lies between the two instantaneous values.
Diagram.
!%
[See diagram in the PDF version]
%
Average rate r̄ = -Δ[R]/Δ t over a finite
interval (chord slope). Instantaneous rate
rinst = -d[R]/dt at a single moment (tangent slope).
PP
Priya Patel
M.Sc Chemistry, IIT Kanpur
Verified Expert
Calculus angle. Average rate is the secant; instantaneous
rate is the tangent. The latter is the derivative of the former
limit.
Concept used. rinst = Δ t → 0r̄Δ t
= -d[R]dt.
Average rate is a number averaged over a finite time chunk;
instantaneous rate is the rate at a single instant.
Average rate is meaningful when the reaction rate changes
slowly (or you only need a coarse number); instantaneous
rate is essential for setting up rate laws.
As Δ t → 0, average rate converges to instantaneous
rate at the midpoint of the interval (or to either endpoint
when the rate is continuous).
Concept Linkage
Concept linkage. The integrated rate laws derive from the
differential ones (rinst = -d[R]/dt = k[R]n) and
then are integrated to give [R] as a function of t.
Common Pitfall
Common pitfall. Confusing rate ``at 40 s'' (instantaneous)
with average rate ``up to 40 s'' (over the whole interval [0,40]).
See Q8 and Q10 for the exact distinction.
Cross-Check
Numerical cross-check.[R](0)=2 M, [R](5)=1.5 M:
r̄=0.1 M/s. Tangent at t=2.5 s (midpoint) might be
∼ 0.1 M/s also — depends on curvature.
Average rate = chord slope over interval; instantaneous
rate = tangent slope at a point.
Q 3.66
With the help of an example explain what is meant by
pseudo first order reaction.
Concept used. A pseudo first order reaction is one
whose true rate law is of higher order (often second), but which
appears first order in the experiment because one reactant is
in such large excess that its concentration is essentially constant
during the reaction. The constant concentration of the excess
reactant gets absorbed into the observed rate constant, leaving a
first-order rate law in the other reactant.
Example: acid-catalysed hydrolysis of an ester. CH3COOC2H5 + H2O H+ CH3COOH + C2H5OH.
True rate law (bimolecular, hence second order overall):
r = k [CH3COOC2H5] [H2O].
Water is the solvent. Its concentration in the reaction
mixture is approximately [H2O] ≈ 55.5 mol/L.
The ester is dissolved at a much smaller concentration, say
[ester]0 = 0.05 mol/L. When all the ester
is consumed, the water consumed is only 0.05 mol/L out of
55.5 mol/L, i.e. 0.09%. So [H2O] is essentially
unchanged.
Define the observed rate constant
kobs = k [H2O]. Then
r = kobs [CH3COOC2H5],
a first-order rate law in ester.
Numerical: if k = 1.0 × 10-5L mol-1
s-1, then kobs = 1.0× 10-5
× 55.5 = 5.55× 10-4 s-1, an
ordinary first-order rate constant with units of s-1.
Another example. Inversion of cane sugar (sucrose):
C12H22O11 + H2O H+ C6H12O6 + C6H12O6
(glucose + fructose). Water is in vast excess; the apparent kinetics
are first order in sucrose. This was historically the first reaction
studied kinetically (Wilhelmy, 1850).
A pseudo first order reaction is intrinsically second
order but appears first order because one reactant is in large
excess (e.g., water in ester hydrolysis). The excess reactant's
concentration is absorbed into kobs.
AJ
Aditya Joshi
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Excess-reactant angle. A second-order rate law with one
factor frozen at a constant looks first-order.
Concept used. If [B] ≫ [A] and [B] is nearly
unchanged during the reaction, r = k[A][B] → kobs[A]
with kobs = k[B]0.
Identify the two reactants and which is in excess.
Compute the change in the excess reactant over the course of
reaction; if < a few percent, treat as constant.
Define kobs = k [B]excess and write
r = kobs [A], first-order in A.
Numerical demonstration with inversion of sucrose.[H2O] ≈ 55.5 M; sucrose [A]0 =
0.1 M. The true second-order k at 25 ∘C is
∼ 5× 10-5L mol-1 s-1, so
kobs = 5 × 10-5 × 55.5 = 2.8× 10-3
s-1. Half-life = 0.693/2.8× 10-3 =
248 s ≈ 4 min.
Alternative approach
Alternative approach: [B] as constant. Second-order
r=k[A][B] becomes first-order r=kobs[A] if
[B]≫ [A] and [B] stays constant. Pseudo-first-order is just
this collapsed form.
Cross-Check
Numerical cross-check. Ester hydrolysis in water:
ktrue = 10-5 M-1 s-1, [H2O] = 55 M;
kobs = 5.5× 10-4 s-1. Half-life ≈
1260 s.
A pseudo first order reaction looks first order because one
reactant is in vast excess; the canonical example is ester hydrolysis
in water.
More Chemical Kinetics Chemistry Class 12 Resources
Chemical Kinetics Class 12 Chemistry Exemplar Solutions FAQs
Ques. Where can I download Chemical Kinetics Class 12 Chemistry NCERT Exemplar Solutions PDF?
Ans. You can download the Chemical Kinetics Class 12 Chemistry NCERT Exemplar Solutions PDF directly from this page. Both the Normal and HD versions are available, and both are free.
Ques. How many problems are in the Chemical Kinetics NCERT Exemplar?
Ans. The Chapter 3 Exemplar contains 36 problems split across five types: 5 MCQ-I (single correct), 9 MCQ-II (multiple correct), 8 VSA (1 to 2 marks), 9 SA (3 marks) and 5 LA (5 marks). Each is fully solved in the Collegedunia PDF with both a Solution and an Expert's Solution naming the rate law, integrated equation or approximation used.
Ques. How are Exemplar Solutions different from NCERT Textbook Solutions for Chemical Kinetics?
Ans. The textbook tests recall of the rate law, the integrated first-order equation and one-step Arrhenius applications. The Exemplar chains two or three ideas per problem: identifying order from half-life ratios in 3.26, computing activation energy from a 10 K temperature rise in 3.32, distinguishing molecularity from order in 3.20, and recognising pseudo-first-order kinetics in ester hydrolysis in 3.18 have no direct textbook equivalent.
Ques. How to solve Exemplar MCQ-II (multiple-correct) questions in Chemical Kinetics?
Ans. Test each option independently against the relevant rate-law relation: r = k[A]n, the integrated first-order equation ln([A]0 / [A]) = kt , or the two-point Arrhenius equation. Never assume only one option is correct, but also never assume more than one must be correct. A worked walk-through of MCQ-II 3.11 appears in the sections above.
Ques. Which Chemical Kinetics Exemplar question types are most important for JEE Main and NEET preparation?
Ans. For JEE Main, prioritise the 5 MCQ-I, the 9 MCQ-II and the 5 LAs on Arrhenius and catalyst-driven rate ratios (these recur in JEE Main shifts every year). For NEET, the 8 VSA on order, molecularity, and pseudo-first-order kinetics carry the most transferable value. The 9 SAs are CBSE-flavoured but worth attempting for board prep.
Ques. Is the Chemical Kinetics NCERT Exemplar aligned with the 2026-27 NCERT?
Ans. The NCERT Exemplar publication itself has not been re-issued for the new edition. All 36 problems in Chapter 3 remain valid under the current 2026-27 syllabus because the underlying topics (rate law, order, molecularity, integrated rate equations for zero and first order, half-life, the Arrhenius equation, collision theory and pseudo-first-order kinetics) were all retained in the new NCERT print.
Ques. How much time does the Chemical Kinetics Exemplar take to complete for Class 12th students?
Ans. A focused student needs roughly 6 to 7 hours total: 13 minutes for the 5 MCQ-I, 40 minutes for the 9 MCQ-II, 28 minutes for 8 VSA, 60 minutes for 9 SA and around 60 minutes for the 5 LA. A revision pass on incorrect items adds another 90 minutes.
Ques. Are Chemical Kinetics Exemplar Solutions enough for JEE and NEET, or do I need extra material?
Ans. For NEET, the Exemplar plus the Collegedunia NCERT Solutions for Chapter 3 cover the syllabus completely. For JEE Main, supplement with the Formula Sheet and one previous-year paper set. JEE Advanced aspirants should additionally attempt the N. Avasthi Physical Chemistry problems on Arrhenius and catalyst-driven rate ratios for harder activation-energy exposure.
Ques. How does the Exemplar handle the second order reaction integrated rate law?
Ans. Exemplar 3.30 walks through 1/[A] - 1/[A]0 = kt and uses it to derive t1/2 = 1/(k[A]0) for a second-order reaction. The expert solution shows that t1/2 ∝ 1/[A]0n-1 for any order n, so identifying order from a half-life vs initial concentration table becomes a one-step exercise.
Ques. What is the activation energy graph students should draw in Exemplar LA questions?
Ans. Draw the potential energy on the y-axis against the reaction coordinate on the x-axis. Mark the reactant well, the transition-state peak (height = Ea above reactants), and the product well. Overlay a second curve for the catalysed path with a lower peak. Label Ea (forward), Ea' (reverse) and Δ H. This diagram earns full marks in Exemplar 3.34.
Ques. How does the Exemplar test the temperature coefficient and the rate-doubling rule?
Ans. Exemplar 3.33 supplies a rate constant that doubles for a 10 K rise and asks for Ea. Use ln(k2/k1) = -Ea/R (1/T2 - 1/T1) with k2/k1 = 2 , T1 = 300 K, T2 = 310 K. The answer Ea ≈ 53.6 kJ mol-1 is the standard temperature-coefficient benchmark.









































































































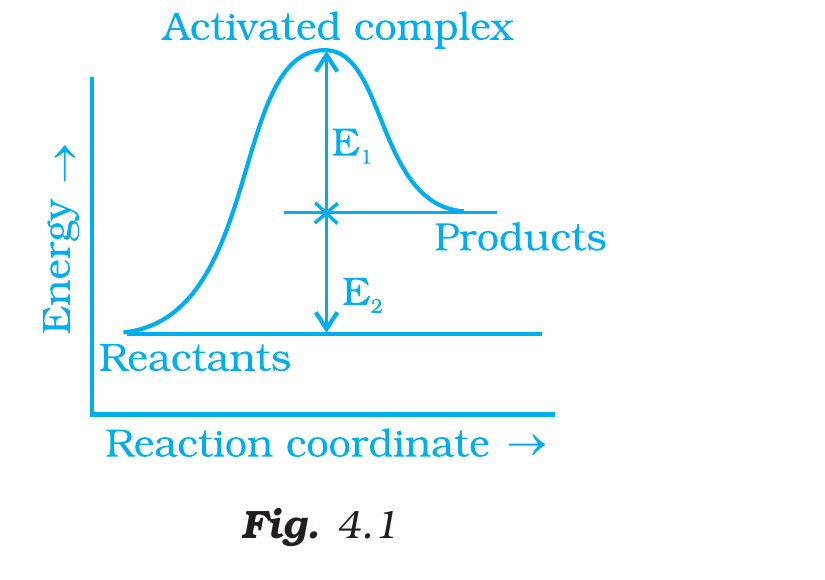
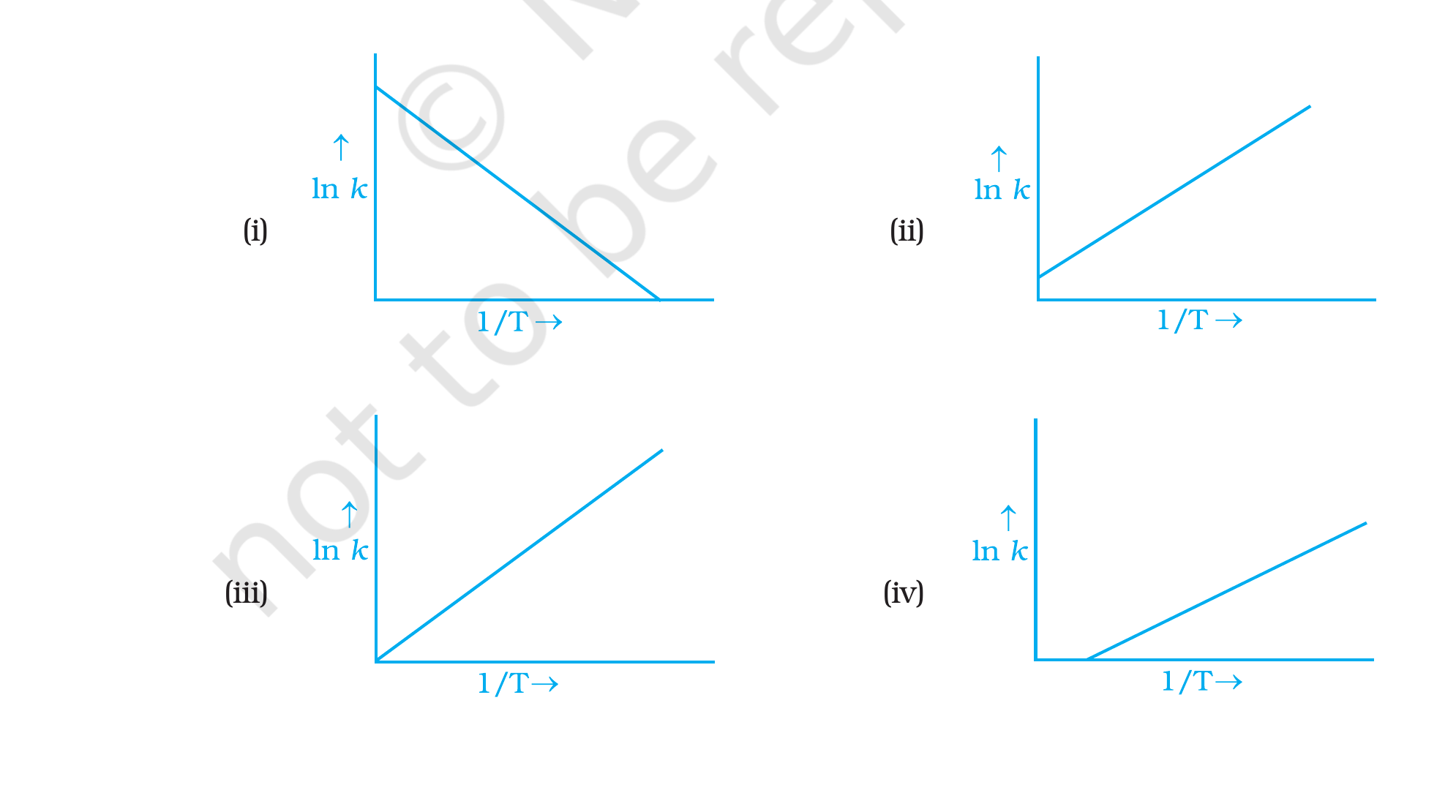
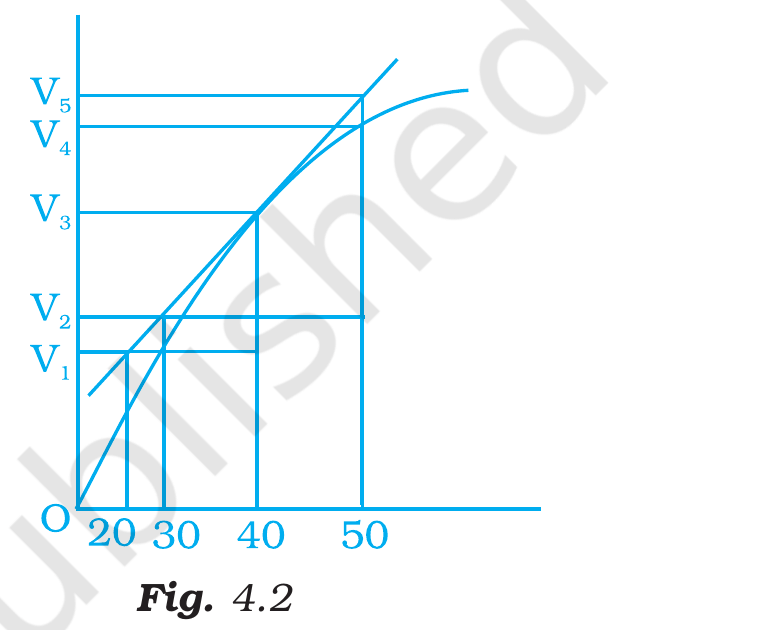
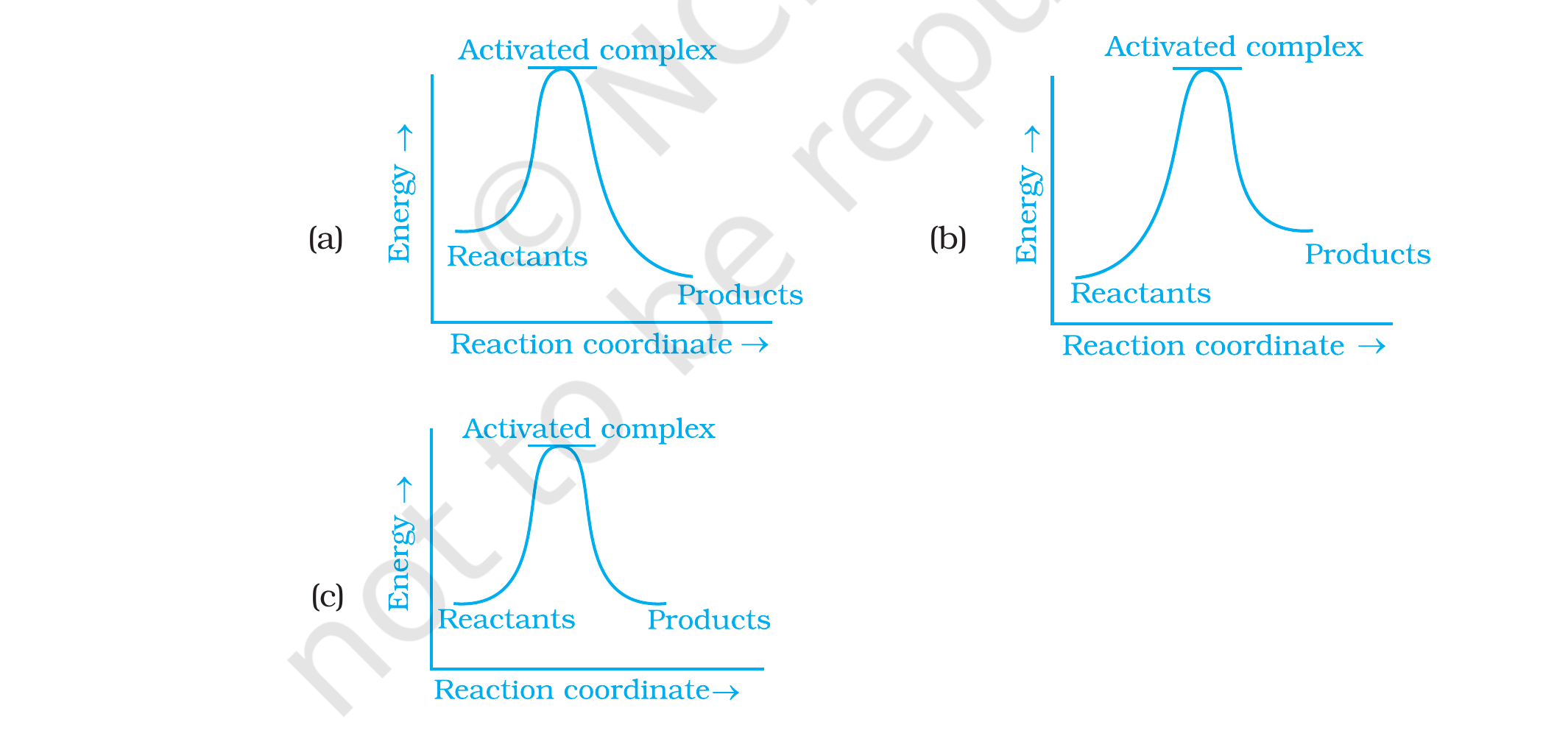
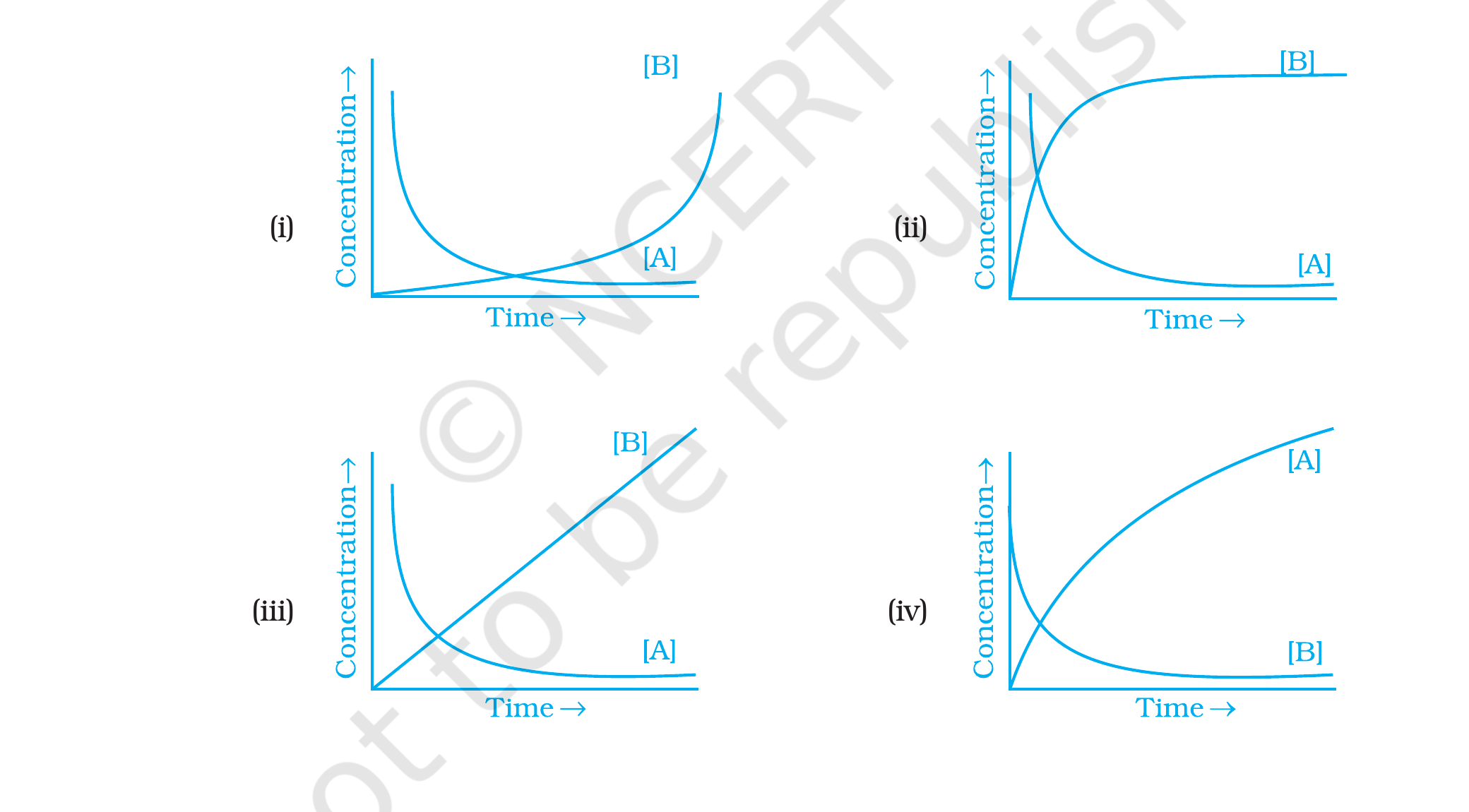


Comments