Class 12 Biology Chapter 9 Biotechnology: Principles and Processes is the chapter that builds the molecular toolkit used in every applied chapter that follows, from restriction enzymes and palindromic recognition sites to PCR thermal cycling and bioreactor scale-up. The 2026-27 NCERT keeps every sub-topic intact, and this 72-page Exemplar Solutions PDF works through all 38 problems mapped to the latest syllabus and the last five NEET keys.
CBSE Weightage: 5 to 7 marks (typically one 2-marker on restriction enzymes plus a long answer on the nine-step rDNA workflow or PCR)
JEE Main Weightage: Not in JEE Main syllabus
NEET Weightage: 3 to 5 questions per year
Chapter 9 Biotechnology: Principles and Processes Exemplar Solutions PDF
Student Pulse: Chapter 9 Biotechnology: Principles and Processes Difficulty Read from a Recent Class 12 Biology Survey
In a recent independent survey of 14,800 Class 12 Biology students conducted before the 2026 boards, 75% rated the PCR step-by-step amplification diagram as the hardest sub-topic in the chapter, even though it routinely carries the highest single-question marks in CBSE and NEET papers.
The same survey gave us the breakdown below, which a Class 12 student should look at before deciding how to allocate revision time across biotechnology: principles and processes class 12 biology exemplar solutions topics.
What 14,800 students told us about the Chapter 9 Biotechnology: Principles and Processes NCERT Exemplar Solutions journey:
75% of students surveyed marked the PCR step-by-step amplification diagram as the hardest sub-topic.
67% reported losing 1-2 marks on naming restriction enzymes and their recognition sequences, even when the rest of their answer was correct.
4 out of 5 students said the gel-electrophoresis labelled apparatus was the most-skipped figure in their answer sheet.
Average student took 6.5 hours for the first read of the chapter, and 2.8 hours for a focused revision pass before the board exam.
Of the 14,800 students surveyed, only 30% attempted all 10 NCERT exercise questions; the rest stopped earlier. Toppers, however, reported attempting every question and revisiting wrong attempts within 24 hours.
Source: 2025-26 Class 12 Biology student survey. Sample of 14,800 students from CBSE-affiliated schools across 18 states.
38 Exemplar problems | 12 MCQ + 5 MCQ-II + 9 VSA + 8 SA + 4 LA | Tools of rDNA, processes of rDNA, bioreactors, downstream · Class 12 Biology Chapter 9, 2026-27 NCERT
These Exemplar Solutions are curated by NEET-rank-holder mentors at Collegedunia, mapped strictly to the 2026-27 NCERT chapter, and benchmarked against the last five years of CBSE Board and NEET papers.
Why Biotechnology Exemplar Practice Decides Your NEET Biology Score
Biotechnology is a 5-to-7 mark CBSE chapter, yet NEET 2024 and NEET 2025 each carried 4 to 5 questions from it, several as assertion-reason items where wrong phrasing scored zero. The chapter rewards exact terminology, the difference between endonuclease and exonuclease, between palindrome and any random repeat, between a plasmid and a cosmid, and the Exemplar is the only place where this terminology is drilled question-by-question. Working all 38 problems in this PDF gives you the recall scaffold that NEET examiners reuse year after year.
How Will Collegedunia's Exemplar Solutions Help You Crack Class 12 Biotechnology?
Biotechnology rewards precise phrasing more than almost any other Class 12 Biology chapter; NEET answer keys reject "ligase joins DNA" written without "phosphodiester bond" and award only the formal mechanism word. Every Exemplar item below carries a full Solution plus an Expert's Solution that names the exact recall phrase the key wants.
Every Question Type Worked End-to-End: all 12 MCQ, 5 MCQ-II (multi-correct), 9 VSA, 8 SA and 4 LA problems with reasoning written out, no skipped steps.
Concept Stack Named: each step lists the principle invoked, whether the EcoRI palindrome rule, the pBR322 insertional-inactivation logic, or the PCR cycle stoichiometry.
NEET Bridge: items are tagged with the NEET year that reused the scaffold so you know which Exemplar problems are highest-yield revision.
2026-27 Aligned: every solution flags that the underlying topic still appears in the current syllabus (no trims for Ch 9).
Sample MCQ Walk-Through: The Most-Missed Restriction-Enzyme Item
MCQs on restriction enzymes pair an enzyme name with a recognition site; decoding the Roman numeral is the bit most students skip. The walk-through below shows the full nomenclature derivation Collegedunia mentors recommend.
Exemplar MCQ: The name EcoRI tells you the enzyme comes from:
(a) Erwinia coli, RY13 strain, first enzyme (b) Escherichia coli, RY13 strain, first enzyme (c) Escherichia coli, R-restriction system, type I (d) Escherichia coli, recombinant DNA, isolate 1
Step 1 - Decode 'Eco'. First letter of genus + first two of species = E (Escherichia) + co (coli). So 'Eco' ⇒ Escherichia coli.
Step 2 - Decode 'R'. Strain code follows. E. coli strain RY13 is the source; only the 'R' is retained in the enzyme name.
Step 3 - Decode 'I'. Roman numeral = order of isolation. EcoRI = the first restriction enzyme isolated from E. coli RY13.
Step 4 - Eliminate distractors. Option (a) wrong genus; (c) wrong meaning of R; (d) wrong meaning of I. Correct: (b).
Biotechnology Principles and Processes Exemplar Question-Type Distribution
Type
Count
Average Marks
NEET Yield
MCQ (single correct)
12
1 each
Very high (most direct-recall NEET)
MCQ-II (multiple correct)
5
1 each
High (assertion-reason source)
VSA (Very Short Answer)
9
1 each
High
SA (Short Answer)
8
2 to 3 each
Medium
LA (Long Answer)
4
5 each
CBSE Boards LA staple
The 12 MCQs + 5 MCQ-II + 9 VSA stack (26 items) is the NEET-prep core; the 8 SA + 4 LA stack (12 items) is the CBSE-Boards core. Solve the NEET core on Day 1.
Sample SA Walk-Through: Why Eukaryotes Lack Restriction Enzymes (Exemplar Q-SA-5)
Exemplar SA: "Do eukaryotes possess restriction endonucleases? Give reasons."
Step 1 (1 mark) - State the answer. No, eukaryotic cells do not possess restriction endonucleases.
Step 2 (1 mark) - Reason 1. Eukaryotic DNA is protected within the nuclear envelope; foreign viral DNA cannot easily invade without first being routed through the nuclear pore. The selective pressure that drove the restriction-modification system in bacteria is therefore absent.
Step 3 (1 mark) - Reason 2. Eukaryotic DNA carries extensive cytosine methylation (CpG islands), so an enzyme that cut unmethylated foreign DNA would risk cutting host DNA at unmethylated sites. Eukaryotes use other anti-viral systems (interferons, RNAi).
Expert's note: CRISPR-Cas9 is sometimes called a "restriction system" in popular media; CBSE explicitly does not treat CRISPR as a restriction endonuclease at Class 12. Stick to the NCERT phrasing.
Common Errors NEET Aspirants Make in Biotechnology Exemplar Questions
Error 1. Writing the enzyme name with an Arabic '1' (EcoR1) instead of Roman 'I' (EcoRI). Single-mark MCQ trap.
Error 2. Confusing the heat-shock temperature (42 degree C) with the PCR annealing temperature (~55 degree C).
Error 3. Calling pBR322 a virus. It is a plasmid.
Error 4. Writing "DNA ligase joins DNA" without the phrase phosphodiester bond. CBSE 2024 docked marks.
Error 5. Spelling Thermus aquaticus as Termus aquaticus. Direct NEET MCQ trap on the source of Taq.
Sample LA Walk-Through: Nine-Step rDNA Workflow (Exemplar Q-LA-2)
The single highest-yield CBSE long-answer (5 marks) from this chapter. The walk-through below names every step with its NCERT phrase.
1. Isolation of DNA. Lysozyme (bacteria) / cellulase (plant) / chitinase (fungi) breaks the wall; SDS lyses the membrane; chilled ethanol precipitates the DNA as fine threads.
2. Cutting at specific locations. Restriction enzymes cut both the foreign DNA and the vector at the same palindrome to generate complementary sticky ends.
3. Amplification by PCR. 25 to 35 cycles of denaturation (94-98 degree C) → annealing (~55 degree C) → extension (72 degree C with Taq) increase the target by ~109.
4. Ligation. DNA ligase forms a phosphodiester bond between the 5'-phosphate of the insert and the 3'-OH of the vector.
5. Insertion of rDNA into host. CaCl2-treated competent E. coli + 42 degree C heat-shock for 90 s; or biolistics for plants; or electroporation for animal cells.
6. Selection of transformants. Selectable marker (antibiotic resistance) lets recombinants grow on selective media; insertional inactivation or blue-white screen distinguishes recombinants.
7. Culture of recombinant clone. Inoculate into bioreactor with sterile medium, controlled pH, temperature and O2.
8. Expression of gene product. Strong promoter + ribosome binding site drive transcription and translation of the GOI.
9. Downstream processing. Centrifuge → chromatography → formulation → QC. DSP can be 50 to 80 percent of total product cost.
Related Resources for Biotechnology Principles and Processes Class 12 Biology
All NCERT Exemplar Questions for Biotechnology Principles and Processes with Step-by-Step Solutions
Every question of the NCERT Exemplar set for Class 12 Biology Chapter 9 Biotechnology Principles and Processes is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
Multiple-Choice Questions
Q 9.1
Rising of dough is due to:
(a) Multiplication of yeast
(b) Production of CO2
(c) Emulsification
(d) Hydrolysis of wheat flour starch into sugars.
Correct option: (b) Production of CO2.
Concept used. Bread dough is fermented by the budding yeast
Saccharomyces cerevisiae, also known as baker's yeast. Under
anaerobic conditions inside the kneaded dough, yeast metabolises the sugars
released from wheat starch through ethanolic fermentation:
C6H12O6 ->[yeast] 2 C2H5OH + 2 CO2.
The CO2 gas produced gets trapped inside the gluten network of the dough,
inflating it like a balloon. This is what makes the dough rise.
Yeast cells take up glucose (and maltose released by amylases acting on
starch). Under low oxygen, glucose is broken down to pyruvate by
glycolysis, then to ethanol + CO2.
Each glucose molecule yields 2 molecules of CO2. The gas cannot
escape the elastic gluten matrix, so it forms millions of tiny bubbles.
These bubbles expand on warming, almost doubling the volume of the dough.
The ethanol mostly evaporates during baking.
Why the other options are wrong. (a) Yeast does multiply in the dough,
but cell division alone adds negligible volume — the inflation is due to gas
release. (c) Emulsification is the dispersion of one liquid in another and is
irrelevant here. (d) Starch hydrolysis only releases sugars; it doesn't
inflate the dough by itself.
Option (b): Production of CO2.
AI
Aanya Iyer
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Read the question as ``what physical change makes
the dough larger?'' rather than ``what biological agent is responsible?''
Volume can only grow if something pushes the dough outward, and inside a sealed
dough the only candidate is a gas.
Identify the gas. Yeast is anaerobic in the dough, so its fermentation
pathway must end in CO2 plus an organic byproduct (here, ethanol):
C6H12O6 -> 2 C2H5OH + 2 CO2.
Identify where the gas goes. Wheat dough is rich in
gluten (a wheat-protein network) that traps CO2 bubbles.
The bubbles cannot escape, so the dough swells.
Connect to the example contrast: dosa/idli batter has no gluten, so the
gas trap is the rice/dal slurry itself; same mechanism, different scaffold.
Why this matters. The bread-and-yeast story is the oldest documented
biotechnology — humans were running anaerobic Saccharomyces bioprocesses
millennia before they had names for them. NEET 2019 and CBSE 2020 both tested it.
Option (b): Production of CO2.
Q 9.2
Which of the following enzymes catalyse the removal of nucleotides
from the ends of DNA?
(a) endonuclease
(b) exonuclease
(c) DNA ligase
(d) Hind II
Correct option: (b) exonuclease.
Concept used.Nucleases are enzymes that hydrolyse the
phosphodiester bond of a nucleic-acid strand. They fall in two camps:
0pt
Exonucleases: act only at the ends of a DNA strand and
snip nucleotides off one at a time (5' or 3' end, depending on the
enzyme).
Endonucleases: cut within the DNA, often at a specific
sequence (these include restriction enzymes like Hind II).
Match the verb in the question — ``removal from the ends'' — to the
definition of an exonuclease. By definition only an exonuclease can act
from an end.
Eliminate (a): an endonuclease cuts internally, not at the ends.
Eliminate (c): DNA ligase joins two DNA pieces by forming a
phosphodiester bond. It is the opposite reaction.
Eliminate (d): Hind II is a restriction endonuclease; it makes
an internal cut at a specific 6 bp palindrome.
Option (b): exonuclease.
PS
Pranav Sharma
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Quick reading. The word ``ends'' is doing all the work in this stem.
In molecular biology only one class of nucleases is defined by acting at strand
termini, so the answer is fixed before you read any option.
Recall the operational definitions: endo cuts inside the molecule,
exo chews at the ends. ``Lyase, ligase, polymerase'' aren't
cleavage enzymes at all.
Map options to definitions: (a) internal cut, (b) end cut, (c) joining,
(d) restriction enzyme = subclass of endonuclease. Only (b) matches.
Practical context: in rDNA work exonucleases are deliberately
avoided once you have your gene fragment, because they will eat
back the sticky ends needed for ligation.
Why this matters. The very next question in any rDNA experiment
(``would you use an exonuclease?'') depends on this distinction. NEET frequently
asks the same fact in the form of a one-line definition match.
Option (b): exonuclease.
Q 9.3
The transfer of genetic material from one bacterium to another through
the mediation of a viral vector is termed as:
(a) Transduction
(b) Conjugation
(c) Transformation
(d) Translation
Correct option: (a) Transduction.
Concept used. Bacteria exchange DNA by three natural mechanisms:
0pt
Transformation — uptake of free, naked DNA from the
surroundings (discovered by Griffith with Streptococcus pneumoniae).
Conjugation — direct cell-to-cell transfer of DNA through a
sex pilus (Lederberg & Tatum's F+ × F- cross).
Transduction — transfer of DNA from one bacterium to another
packaged inside a bacteriophage (a viral vector). Discovered by
Zinder and Lederberg in Salmonella.
Read the discriminator: ``mediation of a viral vector''. Only
one of the three natural transfer mechanisms uses a virus, namely
transduction.
Eliminate (b) conjugation: needs physical pilus contact, not a virus.
Eliminate (c) transformation: needs free DNA, not a virus.
Eliminate (d) translation: that is the ribosome-mediated synthesis of
protein from mRNA, not a DNA-transfer process at all.
Option (a): Transduction.
KR
Karan Reddy
M.Sc Microbiology, JNU
Verified Expert
Strategic angle. The four options test three different vocabulary
families at once — gene transfer, translation, viral biology. Solve by tagging
each option with its discipline before comparing.
Tag the options: transduction (phage), conjugation (pilus),
transformation (naked DNA), translation (ribosome-mRNA).
Match to the stem's keyword ``viral vector''. The only match is
transduction.
Recall the classic experiment: P22 phage transferring genes between
Salmonella typhimurium strains. The phage packages a fragment of
the donor chromosome by mistake, infects a new cell, and dumps that DNA
inside.
Why this matters. Of the three bacterial DNA-transfer methods,
transduction is the one engineers exploit in modern viral vectors (AAV,
lentivirus) used in human gene therapy.
Option (a): Transduction.
Q 9.4
Which of the given statements is correct in the context of visualizing
DNA molecules separated by agarose gel electrophoresis?
(a) DNA can be seen in visible light
(b) DNA can be seen without staining in visible light
(c) Ethidium bromide stained DNA can be seen in visible light
(d) Ethidium bromide stained DNA can be seen under exposure to UV light
Correct option: (d) Ethidium bromide stained DNA can be seen under
exposure to UV light.
Concept used. DNA is colourless and its absorption peak is at 260 nm
(deep UV) so it is invisible to the naked eye in white light. To see DNA bands
on an agarose gel we stain with ethidium bromide (EtBr): a planar
aromatic molecule that intercalates between adjacent base pairs of
the DNA double helix. Once intercalated, EtBr absorbs UV (∼ 300 nm) and
re-emits as bright orange visible light at ∼ 605 nm
(fluorescence). On a UV transilluminator the DNA bands therefore glow
orange against a dark background.
State the two facts the answer depends on:
(i) DNA + visible light = invisible.
(ii) DNA + EtBr + UV light = orange fluorescence.
Test each option against these facts. Options (a), (b) and (c) all
claim that DNA can be seen in visible light, which contradicts fact (i).
Only option (d) combines the stain (needed for any visualisation) with
the right light source (UV).
Option (d): Ethidium bromide stained DNA can be seen under
exposure to UV light.
RB
Riya Bhat
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Picture-first. Think of the lab moment: gel runs, slides into the
transilluminator, lights go off, UV lamp comes on. The bright orange bands you
photograph are EtBr fluorescing under UV — never under room light.
Eliminate any option that puts DNA + visible light together (a, b, c).
DNA absorbs at 260 nm, which is invisible to humans.
Confirm (d) by mechanism: EtBr intercalates, absorbs UV around 300 nm,
re-emits visible light at 605 nm (Stokes shift).
Note the safety caveat: EtBr is a mutagen, UV is harmful to skin and
eyes. The gel is photographed through a UV-blocking filter.
Why this matters. Every gel image you see in a research paper relies
on this trick. Newer SYBR Green and GelRed stains use the same intercalation
+ fluorescence principle.
Option (d): EtBr-stained DNA under UV.
Q 9.5
`Restriction' in Restriction enzyme refers to:
(a) Cleaving of phosphodiester bond in DNA by the enzyme
(b) Cutting of DNA at specific position only
(c) Prevention of the multiplication of bacteriophage by the host bacteria
(d) All of the above
Correct option: (c) Prevention of the multiplication of bacteriophage
by the host bacteria.
Concept used. The word restriction is historical, not
mechanistic. In the 1950s, Salvador Luria observed that phages grown on one
E. coli strain could not infect another strain — their growth was
restricted. The molecular cause, discovered by Werner Arber, Hamilton
Smith and Daniel Nathans (Nobel 1978), is a bacterial defence system: enzymes
that recognise and chop foreign (phage) DNA at specific palindromic sequences
while sparing the host's own DNA, which is protected by methylation. So the
cut-action is the mechanism, but restriction is what the bacterium
achieves by it — restricting phage multiplication.
Distinguish action from purpose. Phosphodiester cleavage (a) and
sequence-specific cutting (b) are the action of the enzyme.
``Restriction'' refers to the purpose it serves the bacterium.
That purpose is to chop incoming bacteriophage DNA into harmless pieces
before it can hijack the host. The host therefore restricts (prevents)
phage multiplication.
Option (d) is wrong because it lumps together the mechanism and the
purpose under one label. Only (c) captures the historical meaning.
Option (c): Prevention of phage multiplication by the host
bacteria.
AV
Aditya Verma
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. Treat this like a vocabulary question: the term
``restriction'' is a noun describing the outcome for the bacterium, not
the chemistry of the cut. So the right answer must talk about phages losing,
not bonds breaking.
Recall Luria's original observation: phage λ grows on
E. coli strain K but not on strain B — strain B restricts
the phage. The molecular explanation came later.
Map this onto the options. (a) and (b) describe the enzyme's bond
chemistry; (c) describes the biological outcome (= restriction).
Eliminate (d): combining purpose with mechanism into ``all of the
above'' would make the term ``restriction'' mean three different things,
which it does not.
Why this matters. The same restriction-modification system inspired
the recent CRISPR-Cas revolution, which is also a bacterial defence
against phages that scientists rerouted into a gene-editing tool.
Option (c): host prevents phage multiplication.
Q 9.6
Which of the following is not required in the preparation of a
recombinant DNA molecule?
(a) Restriction endonuclease
(b) DNA ligase
(c) DNA fragments
(d) E.coli
Correct option: (d)E. coli.
Concept used. A recombinant DNA molecule is built
in vitro by combining a fragment of foreign DNA with a vector
(usually a plasmid). The minimum kit on the bench is:
0pt
A restriction endonuclease to cut both the vector and the
foreign DNA at the same recognition sequence, producing compatible
sticky ends.
A DNA ligase to seal the two phosphodiester gaps that hold the
fragments together.
The DNA fragments themselves — the gene of interest and the
linearised vector.
E. coli is the host cell used to propagate (multiply)
the recombinant molecule after it has been assembled. It is not needed
during the in-vitro ligation step.
Separate the two stages of rDNA work — molecule construction
(in vitro) versus molecule propagation (inside a host).
The question asks what is not required for construction. Map
each option: (a), (b), (c) all sit on the bench, (d) sits in the
incubator.
So E. coli is the odd one out.
Option (d): E. coli.
SJ
Sneha Joshi
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The question is really asking ``which item belongs to
a later step of the workflow?''. Sort the four items chronologically and the
odd one out leaps out.
Chronological order: DNA fragments → restriction enzyme cuts →
ligase joins → recombinant DNA exists. E. coli only
enters after that.
Confirm by definition: ``preparation of a recombinant DNA molecule'' is
an in vitro chemistry step. A bacterial host doesn't participate
in any chemical bond-forming step here.
Eliminate the distractors: even if a student knew E. coli is
the standard host, ``required for preparation'' fixes the timing as
``before transformation''.
Why this matters. This timing distinction shows up again in
insertional inactivation (a post-cloning selection step) and
in PCR (an entirely host-free amplification step).
Option (d): E. coli.
Q 9.7
In agarose gel electrophoresis, DNA molecules are separated on the
basis of their:
(a) Charge only
(b) Size only
(c) Charge to size ratio
(d) All of the above
Correct option: (b) Size only.
Concept used. In agarose gel electrophoresis, all DNA
molecules carry essentially the same charge per unit length — every
nucleotide contributes one negatively charged phosphate group. So the
electric force on every fragment per unit length is the same. What varies
between fragments is how easily they can squeeze through the porous agarose
matrix: smaller fragments wriggle through quickly, larger ones get held back.
The result is that fragments migrate at a rate that depends almost entirely
on their size.
Confirm uniform charge density: DNA backbone = alternating sugar and
phosphate; one phosphate per nucleotide ⇒ uniform
charge-to-mass ratio. So charge alone cannot discriminate.
Confirm size dependence: the agarose matrix acts as a sieve. Migration
distance ∝ 10(1/size in bp) over the linear range.
Compare options: (a) wrong (same charge density), (c) wrong (the ratio
is roughly constant for DNA), (d) wrong (it includes a, c), (b) right.
Option (b): Size only.
AS
Aarav Singh
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Reverse-engineer the answer from the design of the
experiment. If electrophoresis sorted DNA by charge, all DNA would migrate
together (uniform charge density). So the only useful sorting variable left
is size.
Recall the formula for electrophoretic mobility:
μ = qf where q is net charge and f is the frictional
drag from the gel matrix.
For DNA, q scales linearly with length, but so does f in the
sieving regime — except that small molecules sneak through pores
faster, breaking the linear scaling in their favour.
Hence the separation is dominated by sieving, i.e. by molecular size.
Why this matters. The same logic explains why SDS-PAGE
separates denatured proteins by size — SDS uniformly coats them with negative
charge, so the gel sieves by size alone. Same principle, different molecule.
Option (b): Size only.
Q 9.8
The most important feature in a plasmid to serve as a vector in gene
cloning experiment is:
(a) Origin of replication (ori)
(b) Presence of a selectable marker
(c) Presence of sites for restriction endonuclease
(d) Its size
Correct option: (a) Origin of replication (ori).
Concept used. A cloning vector must possess four features:
0pt
Origin of replication (ori) — the DNA sequence where
host DNA polymerase initiates replication. Any piece of DNA linked to
an ori will be replicated by the host machinery.
Selectable marker — usually an antibiotic-resistance gene, to
identify host cells that have taken up the vector.
Cloning sites — unique restriction sites where the foreign
DNA can be inserted.
Small size — easier to manipulate in vitro.
All four matter, but without an ori, the vector cannot replicate, so
the foreign gene cannot be multiplied — defeating the whole purpose of cloning.
Hence ori is the most important.
Define the purpose of cloning: make many identical copies of a gene
inside a host cell. ``Many copies'' requires replication.
Without an ori, the vector is a dead piece of DNA inside the
cell — it will be diluted out as the cell divides.
Selectable marker, cloning sites and small size are practical
conveniences; they don't determine replicability.
Option (a): Origin of replication (ori).
VN
Vivaan Nair
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. Decide what is necessary versus what is
useful. Marker and cloning sites are useful selection/insertion tools,
but without replication the vector cannot persist.
Apply a thought experiment: take a plasmid, remove its ori.
After one host cell division the vector is split between two daughters;
after 20 divisions the original copy is 1/220 of the population —
essentially gone. The gene was never cloned.
Repeat the experiment removing the marker instead: the plasmid still
replicates fine, you just can't easily tell which cells have it.
The clone exists.
Same test for cloning sites: without them you can't insert the gene
at all, but the vector itself still replicates. So the
deal-breaker is the ori.
Why this matters. Every modern shuttle vector carries two ori's
(one for E. coli, one for the eukaryotic host) — proof that the entire
design starts from replication.
Option (a): Origin of replication (ori).
Q 9.9
While isolating DNA from bacteria, which of the following enzymes is
not required?
(a) Lysozyme
(b) Ribonuclease
(c) Deoxyribonuclease
(d) Protease
Correct option: (c) Deoxyribonuclease.
Concept used. The goal of DNA isolation is to liberate
intact DNA from the cell while removing every other macromolecule.
The roles of the four enzymes:
0pt
Lysozyme — digests peptidoglycan in the bacterial cell wall,
cracking the cell open.
Ribonuclease (RNase) — chops up RNA, which would otherwise
co-purify with DNA.
Protease — digests cellular proteins, including DNA-binding
histone-like proteins and contaminating enzymes.
Deoxyribonuclease (DNase) — chops up DNA. Adding it would
destroy the very molecule we are trying to isolate!
Re-state the aim: keep DNA whole, remove everything else.
Match each enzyme to a class of molecules to remove (cell wall,
proteins, RNA) — three are useful.
DNase removes DNA, which is the exact opposite of what we want. Hence
it is the one enzyme that must never be added.
Option (c): Deoxyribonuclease.
DP
Diya Pillai
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Picture-first. Visualise the test-tube at the end of the isolation:
you want a long, viscous, fibrous precipitate of DNA when you add cold
ethanol. Anything that shreds DNA into nucleotides would dissolve that
precipitate.
Sort the enzymes by what they cut: wall (lysozyme), proteins (protease),
RNA (RNase), DNA (DNase).
Cross out the one that cuts DNA — that's DNase.
Sanity check: after lysozyme + protease, the cell contents are
liberated. After RNase, RNA is digested. The DNA is then precipitated
with chilled ethanol and spooled out.
Why this matters. The same logic applies to plant DNA isolation
(swap lysozyme for cellulase) and to fungal DNA (chitinase).
The remove-everything-except-DNA strategy is universal.
Option (c): Deoxyribonuclease.
Q 9.10
Which of the following contributed in popularising the PCR
(polymerase chain reactions) technique?
(a) Easy availability of DNA template
(b) Availability of synthetic primers
(c) Availability of cheap deoxyribonucleotides
(d) Availability of `Thermostable' DNA polymerase
Correct option: (d) Availability of `Thermostable' DNA polymerase.
Concept used. PCR cycles repeatedly between three temperatures:
denaturation at ∼ 95 ∘C (to separate the strands), annealing
at ∼ 55 ∘C (primers bind), extension at ∼ 72 ∘C
(polymerase synthesises). The denaturation step destroys ordinary
DNA polymerases (e.g. E. coli polymerase I). Mullis's original PCR
required adding fresh polymerase after every cycle — slow, expensive,
labour-intensive. The breakthrough was Taq polymerase, isolated from
the hot-spring bacterium Thermus aquaticus, which survives the
∼ 95 ∘C denaturation step. One dose of Taq lasts the whole
30-cycle reaction.
Identify the bottleneck before Taq: the polymerase had to be refreshed
every cycle, making PCR impractical.
Identify the fix: a heat-stable polymerase that survives 95 ∘C.
Taq, with optimum ∼ 75 ∘C, fits.
Eliminate (a)–(c): template DNA, primers and dNTPs were already
available before PCR became routine. They were enabling, but not
popularising.
Option (d): Availability of `Thermostable' DNA polymerase.
IB
Ishita Banerjee
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. Read ``popularising'' as ``what removed the biggest
practical barrier?'' Templates, primers and dNTPs were never the limiting
reagent — labour and time were.
List the cost-driver of pre-Taq PCR: one fresh aliquot of polymerase
per cycle × 30 cycles per reaction × hundreds of
reactions per project = unworkable.
Introduce Taq: one aliquot per reaction, ∼30× cheaper
in enzyme alone, ∼30× faster (no pipette break).
Combine with the thermocycler (programmable heating block)
and PCR went from heroic to routine.
Why this matters. Kary Mullis got the 1993 Nobel for inventing PCR,
but the technique only became universal after Taq was added. Engineering a
bottleneck out of a workflow is often a bigger story than the original
invention.
Option (d): Thermostable DNA polymerase.
Q 9.11
An antibiotic resistance gene in a vector usually helps in the
selection of:
(a) Competent bacterial cells
(b) Transformed bacterial cells
(c) Recombinant bacterial cells
(d) None of the above
Correct option: (c) Recombinant bacterial cells.
Concept used. In modern rDNA technology vectors carry two
antibiotic-resistance genes flanking the cloning site (e.g. pBR322
carries ampR and tetR). When the foreign DNA is
inserted into one of these genes (say tetR) it is
inactivated by insertion — the gene no longer works. So:
0pt
Cells that picked up the recombinant plasmid are
ampR but tet-sensitive.
Cells that picked up the empty self-ligated plasmid are still
ampR tetR.
The differential antibiotic response is what lets us select the
recombinants from the non-recombinants. (Mere transformation is selected
on the first antibiotic alone.)
Recall the standard pBR322 selection: plate on ampicillin first to
get all transformants, then replica-plate onto tetracycline. Colonies
that grow on amp but die on tet are the recombinants.
Note that the question says ``usually helps in the selection of'' —
this matches the recombinant-selection use case.
Eliminate (a) competent cells: those are made chemically, not selected
by antibiotic. Eliminate (b) transformed cells: that takes one
antibiotic, but the differential value of a second marker is
recombinant selection.
Option (c): Recombinant bacterial cells.
TM
Tara Mehta
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The verb ``selection'' triggers a workflow question:
at which step are we using the antibiotic? Plate-1 (any transformant survives)
or plate-2 (only recombinants survive)? The marker's discriminating power
shows up on plate-2.
Step 1 (transformation selection): grow on ampicillin. All cells
carrying the plasmid (recombinant or not) live. Cells without the
plasmid die.
Step 2 (recombinant selection): replica-plate on tetracycline.
Recombinants (with the insert in tetR) die on tet;
non-recombinants live. We pick the tet-sensitive ampicillin
survivors → recombinants.
So the two-antibiotic design's purpose is recombinant selection,
which is option (c).
Why this matters. The newer blue-white screening replaces
the second antibiotic with a colour reaction (X-gal), but the logic is
identical: pick the colony where the insertion broke the marker.
Option (c): Recombinant bacterial cells.
Q 9.12
Significance of `heat shock' method in bacterial transformation is to
facilitate:
(a) Binding of DNA to the cell wall
(b) Uptake of DNA through membrane transport proteins
(c) Uptake of DNA through transient pores in the bacterial cell wall
(d) Expression of antibiotic resistance gene
Correct option: (c) Uptake of DNA through transient pores in the
bacterial cell wall.
Concept used. Bacterial cells do not normally take up DNA. To make
them competent, we incubate them on ice with divalent
Ca^2+ ions, which neutralise the negative charges on both the cell
wall and the DNA so they no longer repel each other and the DNA sticks to the
cell surface. A brief heat shock (42 ∘C for ∼ 90 s,
then back to ice) creates transient pores in the wall through which
the surface-bound DNA slips inside. Without the heat shock the DNA sits on the
outside; without Ca^2+ it never gets close enough to slip in.
Recall the three-step competent-cell protocol: (i) ice-cold
CaCl2 treatment, (ii) DNA addition + further ice incubation,
(iii) 42 ∘C heat shock for 90 s, (iv) recovery in growth
medium.
The heat shock physically perturbs the cell envelope, opening transient
pores. The temperature gradient (ice → warm → ice) is what
drives the perturbation.
Eliminate (a): binding to wall is the role of Ca^2+, not heat.
Eliminate (b): bacteria have no DNA-import membrane proteins.
Eliminate (d): heat shock doesn't switch on antibiotic genes.
Option (c): Uptake of DNA through transient pores.
RV
Rohit Verma
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Trace the journey of the DNA molecule from outside
the cell to inside. Ca^2+ gets it to the wall. The heat shock is the
push it needs to cross the wall. So the heat shock answers ``how does it get
in?''.
Map cause to effect: heat shock → thermal stress on the lipid
bilayer → transient pores.
Confirm by negative control: skip the heat shock and the
transformation efficiency drops by 2–3 orders of magnitude. Skip
Ca^2+ and it drops by 3–4 orders of magnitude. Both steps are
needed but they do different jobs.
Cross-check the wrong options against this picture. (a) binding is
before pore opening; (b) no transporter exists; (d) gene expression is
a later step (during recovery in medium).
Why this matters. The competent-cell trick is the gateway to every
plasmid-based cloning experiment in undergraduate and research labs. Every
E. coli DH5α stock you ever buy was made competent this way.
Option (c): transient pores in the cell wall.
Q 9.13
The role of DNA ligase in the construction of a recombinant DNA
molecule is:
(a) Formation of phosphodiester bond between two DNA fragments
(b) Formation of hydrogen bonds between sticky ends of DNA fragments
(c) Ligation of all purine and pyrimidine bases
(d) None of the above
Correct option: (a) Formation of phosphodiester bond between two DNA
fragments.
Concept used. A phosphodiester bond is the covalent linkage
between the 3'-OH of one nucleotide and the 5'-phosphate of the next. The
sugar-phosphate backbone of a DNA strand is held together by these bonds. When
a restriction enzyme cuts DNA, it breaks exactly these bonds — leaving free
3'-OH and 5'-phosphate ends. DNA ligase re-seals these breaks
in an ATP-dependent reaction. After two compatible sticky ends base-pair
(via hydrogen bonds, which happen spontaneously), the ligase covalently joins
them with two new phosphodiester bonds (one per strand).
Recall the chemistry of the cut: restriction enzyme breaks the
phosphodiester bond between the sugar and the next phosphate.
Recall the chemistry of the repair: ligase consumes one ATP per
bond, and forms a new phosphodiester bond between 3'-OH and
5'-PO4.
Distinguish from hydrogen-bonding: the H-bonds between sticky ends
form spontaneously without ligase. Ligase's unique job is the
covalent step.
Eliminate (b): H-bonding is base-pairing, not ligation. Eliminate (c):
ligase does not act on bases. Eliminate (d): (a) is correct.
Option (a): Formation of phosphodiester bond between two DNA
fragments.
KG
Krishna Gupta
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. The trick is to separate the two physical processes
that happen at a sticky-end junction: (i) base-pairing (spontaneous,
H-bonds), and (ii) backbone sealing (enzymatic, covalent). Only one of these
needs ligase, and that's the answer.
Draw the junction: two strands lined up sticky-to-sticky. The base
pairs are already H-bonded after annealing, but there are two
nicks (one per strand) — gaps in the sugar-phosphate backbone.
Identify the role of ligase: catalyse formation of phosphodiester bond
across each nick. Reaction: DNA3'-OH +
DNA5'-PO4 ATP, Mg2+
sealed DNA + AMP + PPi.
Cross-check the wrong options: (b) is what happens before
ligase acts (annealing). (c) is nonsense; ligase doesn't touch bases.
Why this matters. Without ligase you'd have only physical pairing of
sticky ends, which falls apart at the next thermal jostle. Ligase makes the
recombinant molecule chemically real.
Option (a): Phosphodiester bond between two DNA fragments.
Q 9.14
Which of the following bacteria is not a source of restriction
endonuclease?
(a) Haemophilus influenzae
(b) Escherichia coli
(c) Entamoeba coli
(d) Bacillus amyloliquefaciens
Correct option: (c)Entamoeba coli.
Concept used. Restriction enzymes are named after the bacterium they
come from: the first letter of the genus, the next two letters of the species,
plus a strain letter and a Roman numeral (e.g. EcoR I from
Escherichia coli RY13). Crucially, restriction enzymes occur in
bacteria as a defence against phage DNA. Entamoeba coli is
not a bacterium; it is a protozoan (a single-celled eukaryote) that
lives in the human gut. So it cannot be a source of restriction enzymes.
Recall the source organisms of common restriction enzymes:
Hind III ←Haemophilus influenzae strain
Rd; EcoR I ←Escherichia coli RY13;
BamH I ←Bacillus amyloliquefaciens H.
Test Entamoeba coli: this is a protozoan (not
to be confused with the pathogen Entamoeba histolytica). It
lacks the restriction-modification defence system that bacteria
evolved.
So three of four options are bacterial; Entamoeba coli is the
misfit.
Option (c): Entamoeba coli.
YK
Yash Kapoor
M.Sc Microbiology, JNU
Verified Expert
Strategic angle. The question is partly a Latin-name trap. Match
each genus to the kingdom: Haemophilus (bacterium), Escherichia
(bacterium), Entamoeba (protozoan), Bacillus (bacterium).
Apply the rule: restriction enzymes are a bacterial weapon
against phages.
Find the non-bacterium in the list: Entamoeba is a protozoan
(eukaryote), so option (c).
Sanity check via well-known enzymes:
Hind III ←Haemophilus,
EcoR I ←Escherichia,
BamH I ←Bacillus. The fourth would have
to be Ent ? — no such named enzyme exists.
Why this matters. The naming convention is itself a memory aid in
NEET — if you can decode the prefix back to a genus, you can pre-narrow MCQ
answers.
Option (c): Entamoeba coli (not a bacterium).
Q 9.15
Which of the following steps are catalysed by Taq DNA polymerase in a
PCR reaction?
(a) Denaturation of template DNA
(b) Annealing of primers to template DNA
(c) Extension of primer end on the template DNA
(d) All of the above
Correct option: (c) Extension of primer end on the template DNA.
Concept used. PCR cycles between three temperatures, and each
temperature does one job; not all three jobs need an enzyme.
0pt
Denaturation (95 ∘C) — heat alone breaks the
H-bonds between the two DNA strands. No enzyme needed.
Annealing (50–60 ∘C) — primers find their
complementary sequences and base-pair spontaneously. No enzyme needed.
Extension (72 ∘C) — Taq polymerase adds
dNTPs to the 3'-OH of each annealed primer, copying the template.
This is the only enzyme-catalysed step.
Pair each PCR step with its driver: denaturation → heat;
annealing → complementary base-pairing; extension →Taq polymerase.
Eliminate (a) and (b): heat and H-bonding don't need a polymerase.
Confirm (c): Taq adds nucleotides at 72 ∘C
(its optimum), so extension is its job.
Option (c): Extension of primer end on the template DNA.
MD
Meera Desai
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Picture-first. Think of the thermocycler block changing colour as
temperatures rise and fall. The enzyme is along for the ride; it only
works at 72 ∘C, doing nothing useful at 95 ∘C
or 55 ∘C.
State Taq's optimum temperature (∼ 72 ∘C).
That matches the extension step.
State Taq's reaction: add a dNTP to a 3'-OH end if it is
Watson-Crick complementary to the templating base. This is
template-directed primer extension.
Conclude: Taq catalyses extension only. The other two PCR
steps are physical, not enzymatic.
Why this matters. The thermostability of Taq also explains
why we get point-mutation errors in PCR — Taq lacks 3'→5'
proofreading. For high-fidelity PCR we use Pfu (with proofreading).
Option (c): Extension of primer end on the template DNA.
Q 9.16
A bacterial cell was transformed with a recombinant DNA molecule that
was generated using a human gene. However, the transformed cells did not produce
the desired protein. Reasons could be:
(a) Human gene may have intron which bacteria cannot process
(b) Amino acid codons for humans and bacteria are different
(c) Human protein is formed but degraded by bacteria
(d) All of the above
Correct option: (d) All of the above.
Concept used. Expressing a human gene in a bacterial host runs into
several mismatch issues:
0pt
Introns — human genes have introns that must be spliced out
by the eukaryotic spliceosome. Bacteria lack splicing machinery, so
they cannot make a functional mRNA from a raw human genomic gene.
Codon usage bias — although the genetic code is universal,
humans and bacteria prefer different synonymous codons. A human gene
loaded with rare-in-E. coli codons translates poorly.
Proteolysis — even if a human protein is produced, the
bacterial proteases may recognise it as foreign and chop it up.
Walk a human gene through bacterial expression:
transcription → no splicing → mRNA still has introns →
non-functional protein. Reason (a) confirmed.
If you supply a cDNA (intron-free): transcription → translation,
but rare codons stall ribosomes → poor yield. Reason (b) confirmed.
If translation succeeds: bacterial proteases may degrade the
foreign protein. Reason (c) confirmed.
All three reasons are valid contributors. Hence (d) is the right answer.
Option (d): All of the above.
SC
Siddharth Chatterjee
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. ``All of the above'' is correct only when each
individual reason is independently true. Apply the central dogma in a
bacterial host (transcription → translation → folded protein) and
check whether each named hurdle is real at its own step. If any one fails,
``all of the above'' would be wrong.
Test reason (a) at the transcription/processing step. Bacteria have
no eukaryotic spliceosome, so introns in a raw human genomic gene are
never excised. The translated product is gibberish.
True — introns block expression of intron-containing human genes.
Test reason (b) at the translation step. Although the genetic code is
universal, the tRNA pool in E. coli is biased towards
bacterial codon preferences. Human genes loaded with rare-in-bacteria
codons cause ribosome stalling and premature termination.
True — codon-usage mismatch lowers yield.
Test reason (c) at the folding/stability step. Bacterial proteases
like Lon, ClpXP and FtsH actively target misfolded or unfamiliar
proteins for degradation. A human protein produced inside
E. coli often looks ``foreign'' enough to be degraded.
True — proteolysis chops the product.
Since all three independent tests return True, ``All of the above''
(d) is the only consistent answer.
Why this matters. This single MCQ summarises why early human-insulin
production in bacteria was a decade-long engineering puzzle, not a simple
chemistry problem. Each fix — cDNA cloning, codon
optimisation, protease-deficient strains — was a separate research
breakthrough.
Option (d): All of the above are independently true reasons
why a human gene may fail to express in E. coli.
Q 9.17
Which of the following should be chosen for best yield if one were to
produce a recombinant protein in large amounts?
(a) Laboratory flask of largest capacity
(b) A stirred-tank bioreactor without in-lets and out-lets
(c) A continuous culture system
(d) Any of the above
Correct option: (c) A continuous culture system.
Concept used. For industrial-scale protein production, three reactor
geometries are possible:
0pt
Batch culture (a closed flask, even a giant one) — nutrients
run out and waste builds up; growth stops.
Stirred-tank batch bioreactor — same problem at industrial
scale; one batch, then clean and restart.
Continuous culture system — fresh medium flows in, used
medium and product flow out, so the cells stay at log phase
(exponentially growing) for days or weeks.
A continuous system keeps the bioreactor permanently in the optimal growth
phase, giving the highest steady-state yield of recombinant protein.
Compare yield over time. A batch flask peaks once, then dies — total
protein per litre is limited by initial nutrient pool.
A continuous system keeps replenishing nutrients and removing waste,
so it produces protein continuously at the cells' maximum rate.
Eliminate (a): flask size cannot fix the batch-culture problem.
Eliminate (b): a closed stirred tank is still a batch reactor.
Eliminate (d): not ``any of the above'' — (c) is strictly better.
Option (c): A continuous culture system.
AK
Ananya Kumar
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Re-frame the question as ``which option keeps the
cells in log phase the longest?'' Log phase is when proteins are produced
fastest, so the answer must be the option that prolongs it.
Plot yield versus time for each option mentally. Batch flask: bell
curve, peaks once. Closed stirred tank: same bell, just bigger.
Continuous: a flat top that goes on for days.
The integral of yield over time is much larger for the continuous
case, even if the instantaneous peak is similar.
Practical engineering bonus: with continuous flow, the downstream
purification rig can be sized for steady throughput, not for sudden
batch surges — overall cheaper per kg of protein.
Why this matters. This is why recombinant insulin and growth hormone
are produced in continuous fed-batch bioreactors, not in flasks. Scale alone
isn't enough — design matters.
Option (c): A continuous culture system.
Q 9.18
Who among the following was awarded the Nobel Prize for the
development of PCR technique?
(a) Herbert Boyer
(b) Hargovind Khurana
(c) Kary Mullis
(d) Arthur Kornberg
Correct option: (c) Kary Mullis.
Concept used.Kary B. Mullis invented PCR in 1983 while
working at Cetus Corporation, and was awarded the 1993 Nobel Prize in
Chemistry (shared with Michael Smith). The other three scientists are famous,
but for different work:
0pt
Herbert Boyer — co-developed the first recombinant DNA molecule (with
Stanley Cohen, 1973); founded Genentech.
Har Gobind Khorana — Nobel 1968 for cracking the genetic code
(translation, not PCR).
Arthur Kornberg — Nobel 1959 for discovering DNA polymerase I
(a different polymerase, not the PCR technique).
Match each name to its primary contribution. Only Mullis matches the
PCR keyword.
Eliminate the distractors by their actual Nobel work.
Option (c): Kary Mullis.
AR
Aditi Rao
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Quick reading. Pure fact recall. The discriminating word is ``PCR
technique''; the only name on the list tied to PCR is Kary Mullis.
Confirm the year and prize: 1993 Nobel in Chemistry, shared with
Michael Smith (who developed site-directed mutagenesis).
Cross-check the distractors against their actual Nobel topics
(recombinant DNA, genetic code, DNA polymerase). None of them won for
PCR.
Why this matters. Naming the inventor of a foundational technique is
a standard NEET/CBSE one-mark question. Memorise the (invention, inventor, year)
triples for PCR, rDNA, RNA interference, CRISPR.
Option (c): Kary Mullis.
Q 9.19
Which of the following statements does not hold true for restriction
enzyme?
(a) It recognises a palindromic nucleotide sequence
(b) It is an endonuclease
(c) It is isolated from viruses
(d) It can produce the same kind of sticky ends in different DNA molecules
Correct option: (c) It is isolated from viruses.
Concept used. Restriction enzymes are produced by bacteria as
a defence against viruses (bacteriophages) — they are not isolated
from viruses. The classic source organisms are
Haemophilus influenzae (Hind III),
Escherichia coli (EcoR I) and
Bacillus amyloliquefaciens (BamH I).
The other three statements are true:
0pt
(a) Type-II restriction enzymes recognise short
palindromic sequences (e.g. EcoRI recognises 5'-GAATTC-3'
which reads the same on the complementary strand).
(b) They cut within DNA (between bases of a recognition site),
so they are endonucleases.
(d) Cutting two different DNAs with the same enzyme yields identical
sticky ends, which is what makes them ligatable to each other — the
very basis of rDNA technology.
Spot the falsehood. Restriction enzymes come from bacteria, not
viruses. They evolved to restrict (prevent) viral infection.
Verify each true statement quickly: palindrome (yes), endonuclease
(yes), uniform sticky ends across substrates (yes — the whole point
of cloning).
So (c) is the statement that does not hold.
Option (c): It is isolated from viruses.
NP
Neha Patel
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The question pretends to ask about properties, but
the trap is in the source organism. Three of four statements are basic
biology of restriction enzymes; only (c) is a swapped fact.
Sort the four statements into ``properties'' (a, b, d) and ``origin''
(c). Property statements are textbook; the origin claim is the
suspect.
Test the origin: bacteria produce them; the role is to chop viral DNA.
So (c) reverses the biology.
Why this matters. The same reverse-the-relationship trick pops up in
many biology MCQs — e.g. ``vaccines are isolated from antibodies'' (false;
they prime the body to make antibodies). Always check the direction of every
relationship.
Option (c): It is isolated from viruses (FALSE).
Very Short Answer Type Questions
Q 9.20
How is copy number of the plasmid vector related to yield of
recombinant protein?
Concept used. The copy number of a plasmid is the average
number of plasmid molecules per host cell. Recombinant protein yield is
roughly proportional to the number of mRNA transcripts the cell can make from
the gene, which in turn depends on how many copies of the gene template are
present. So more plasmid copies → more mRNA → more protein.
State the chain: copy number ∝ gene-template count ∝
mRNA produced ∝ protein yield (assuming translation is not
the bottleneck).
For pUC-type high-copy plasmids (∼ 500 copies per cell) yield
is far higher than for low-copy BAC vectors (∼ 1 copy).
Yield is directly (approximately linearly) proportional to plasmid
copy number.
IM
Ishaan Mehta
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Quick reading. ``Copy number'' is set by the plasmid's
origin of replication. Vectors chosen for protein production carry
high-copy oris precisely to boost yield.
High copy number ⇒ high gene dosage ⇒
proportionally more mRNA, more protein.
Practical limit: too high a copy number stresses the host
(metabolic burden) and can stop cell division. So the
relationship is monotonic but saturates.
Why this matters. Vector choice is a yield-vs-stability trade-off.
For industrial insulin pUC-type origins win.
Copy number ↑ ⇒ yield ↑, until the
metabolic-burden ceiling.
Q 9.21
Would you choose an exonuclease while producing a recombinant DNA
molecule?
Concept used. An exonuclease chews nucleotides off the
ends of a DNA strand. During rDNA construction we deliberately
create ends — the sticky ends produced by restriction enzymes — and
these are exactly what we need to ligate the foreign DNA into the vector.
An exonuclease would chew those ends back, destroying the sticky overhangs
and making ligation impossible.
Identify the ends in play: sticky 5' or 3' overhangs left by the
restriction enzyme on both vector and insert.
If exonuclease acts: overhangs trimmed → blunt or no end →
no complementary base-pairing → no ligation.
No. An exonuclease would destroy the sticky ends needed for
ligation.
PS
Pooja Sharma
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The recombinant strategy depends on preserved
overhangs. An enzyme that erodes ends is the opposite of what we want. Use the
defining-property test: an exonuclease is, by definition, an end-degrader.
Confirm the chemistry: an exonuclease cleaves the terminal
phosphodiester bond at either the 3'-OH or 5'-PO4 end of a
linear DNA strand, releasing one free dNMP per catalytic cycle.
In rDNA work, we use only restriction endonucleases (which cut
at internal palindromes) plus DNA ligase (which re-seals matched
ends). No exonuclease belongs in this kit.
Worked exception: some post-ligation purifications add an
exonuclease (e.g. Plasmid-Safe DNase) deliberately to remove unligated
linear contaminants. Different timing, different purpose.
Why this matters. The same logic — protect the sticky ends at all
costs during construction — explains why ligation buffers are kept on ice and
why molecular biologists never freeze-thaw their cut DNA more than once.
No, never during construction — it would destroy the sticky ends
needed for ligation.
Q 9.22
What does H in d and `III' refer to in the enzyme Hind III?
Concept used.Restriction-enzyme naming is a four-part
code based on the source organism and order of discovery (Smith & Nathans
convention, 1973):
0pt
H — first letter of the genus name
(Haemophilus).
in — first two letters of the species name
(influenzae).
d — the strain from which it was isolated
(H. influenzae strain Rd).
III — Roman numeral for the order of discovery (the
third restriction enzyme purified from that strain).
H =Haemophilus; in =influenzae;
d = strain Rd; III = third enzyme of that strain (Smith & Nathans
convention).
AS
Aanya Singh
M.Sc Microbiology, JNU
Verified Expert
Quick reading. The name itself is a mnemonic of the source biology
that you can decode without any external lookup, which is what makes the
Smith & Nathans convention so useful in modern catalogue databases.
Decompose: H (first letter of genus Haemophilus)
+in (first two letters of species influenzae)
+d (laboratory strain Rd of that species)
+III (Roman numeral for the third restriction enzyme
purified from that strain).
Cross-check with a different example: EcoR I decomposes as
E. coli, strain R (Y13), first enzyme. Same template.
Source organism, strain identifier and discovery order are all
recoverable from the name alone — no separate metadata file needed.
Why this matters. REBASE, the master restriction-enzyme database, is
ordered by these names; learning the convention lets you parse any catalogue
or product list at a glance.
H =Haemophilus, in =influenzae, d = strain Rd,
III = third enzyme purified from that strain.
Q 9.23
Restriction enzymes should not have more than one site of action in
the cloning site of a vector. Comment.
Concept used. A cloning vector is designed so that
exactly one cut by the chosen restriction enzyme linearises the vector
at the cloning site, leaving room to ligate the insert. If the enzyme had
more than one site within the vector, cutting would chop the vector
into multiple pieces:
One useful piece (the backbone with ori and marker) and one or
more unwanted fragments would be released.
Re-ligation in the presence of the insert could produce a chaotic
soup of multiple recombinant forms, most of which are non-functional.
The ori or marker gene might itself be excised, destroying the
vector's ability to replicate or to be selected.
Multiple cut sites would shatter the vector instead of linearising
it, destroying the ori or marker and producing useless ligation
products.
KN
Krishna Nair
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. A clean cloning experiment needs a single,
predictable cut. Two or more cuts break that predictability.
Geometric reasoning: a circular plasmid cut at n sites yields n
linear fragments. With n>1, the backbone is no longer a single
piece, and there is no guarantee the insert ends up between
ori and marker.
This is why every vector has a multiple cloning site (MCS)
designed so the standard enzymes (EcoRI, BamHI, HindIII) each
cut only once within the MCS and nowhere else.
Multiple cuts destroy the vector's integrity; the MCS is engineered
to guarantee single cuts.
Q 9.24
What does `competent' refer to in competent cells used in
transformation experiments?
Concept used. A competent cell is one whose envelope has
been pre-treated so it can take up exogenous DNA from the surroundings.
Bacterial cells are normally impermeable to DNA. After treatment with
ice-cold CaCl2 (which neutralises the negative charges on the cell wall
and on the DNA), the cell becomes competent — i.e. able to be
transformed.
`Competent' means the cell has been chemically prepared (e.g. with
CaCl2) so its envelope is permeable enough to take in foreign DNA during
transformation.
SB
Sanya Bhat
M.Sc Microbiology, JNU
Verified Expert
Quick reading. ``Competent'' is a lab-jargon shorthand for
``DNA-uptake-ready'' — a state engineered into the cells by an explicit
chemical or electrical pre-treatment, not an intrinsic property.
Native bacterial walls repel DNA: both surfaces carry phosphate-based
negative charges. Treatment with Ca^2+ screens those charges,
letting DNA approach the wall and stick.
A subsequent 42 ∘C heat shock for ∼ 90 s
creates transient pores in the cell envelope, through which the
surface-bound DNA enters the cytoplasm.
Electroporation achieves the same end via a sharp electric pulse
instead of heat. Either way the cell is made transiently permeable
(= competent).
Why this matters. Commercial competent-cell preparations (DH5α,
TOP10, BL21 DE3) are sold ready-made at ∼108 transformants per
g of DNA. The competence step is the most efficiency-determining
preparation in cloning.
Cells made permeable to DNA by a chemical (CaCl2) or physical
(electroporation) pre-treatment.
Q 9.25
What is the significance of adding proteases at the time of
isolation of genetic material (DNA)?
Concept used. Cellular DNA is heavily coated with proteins — most
prominently histones (in eukaryotes) and histone-like proteins
(in bacteria), plus countless transcription factors and packaging proteins.
Proteases chop these proteins into amino acids, freeing the DNA
and also destroying nucleases that would otherwise degrade it.
Free DNA from protein scaffold → better recovery.
Destroy contaminating nucleases (which are themselves proteins) →
better DNA integrity.
Proteases strip away DNA-binding proteins and destroy contaminating
nucleases, so the isolated DNA is both pure and intact.
DI
Dev Iyer
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. ``Anything that isn't DNA must go'' is the
isolation mantra. Proteases handle the protein bucket.
RNase removes RNA, protease removes proteins, organic solvents
partition lipids. What is left is pure DNA.
Removes DNA-bound proteins and contaminating nucleases.
Q 9.26
While doing a PCR, `denaturation' step is missed. What will be its
effect on the process?
Concept used.Denaturation at 95 ∘C melts the
double-stranded DNA into single strands. Only single-stranded template can
be bound by the primers in the next (annealing) step. If denaturation is
skipped, the DNA stays double-stranded, primers cannot anneal, and
Taq polymerase has no primer-template junction to extend.
No denaturation ⇒ no single-stranded template.
No template ⇒ no annealing ⇒ no extension.
Net result: zero amplification — no PCR product.
No amplification will occur because the template stays
double-stranded, blocking primer binding and extension.
AR
Aditya Reddy
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Picture-first. Imagine a zipper that needs to be open before a
bookmark can be slipped in. No melting = no zipper opening = no bookmark.
State the role of each step: denature (open strands), anneal (place
primers), extend (build new strand). Each depends on the previous.
Skipping denaturation breaks the very first link of the chain.
Zero amplification — every downstream step is blocked.
Q 9.27
Name a recombinant vaccine that is currently being used in
vaccination programme.
Concept used. A recombinant vaccine is one in which the
antigen is produced by inserting the corresponding gene into a host cell
(yeast or bacterium) and then purifying the protein. The most widely used
recombinant vaccine is the Hepatitis B vaccine: the gene encoding
the hepatitis B surface antigen (HBsAg) is expressed in Saccharomyces
cerevisiae, and the purified antigen is the vaccine.
Hepatitis B vaccine — produced by recombinant HBsAg expression in
yeast.
RG
Rahul Gupta
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Quick reading. The classical example in NCERT and in NEET is the
recombinant Hepatitis B vaccine.
HBsAg gene cloned into yeast → yeast secretes the antigen →
antigen purified and formulated.
Used worldwide since 1986; part of India's Universal Immunisation
Programme.
Recombinant Hepatitis B vaccine.
Q 9.28
Do biomolecules (DNA, protein) exhibit biological activity in
anhydrous conditions?
Concept used. The three-dimensional shape of every biomolecule —
the DNA double helix, the folded protein, the hydrated enzyme active site —
depends on a continuous hydration shell of water molecules that
forms hydrogen bonds with polar groups. Remove the water and the molecule
collapses or unfolds, losing biological activity. This is why dehydrated
seeds, dried blood spots and lyophilised enzymes are stable but inactive
until rehydrated.
No. Biomolecules need water for their active 3D structure and
enzyme catalysis; in anhydrous conditions they are biologically inactive
(though they may be chemically stable).
PB
Priya Banerjee
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. Biological activity = correct 3D shape +
correct dynamics. Both require water.
Water stabilises the major and minor grooves of DNA via hydration of
phosphates. Without water, the helix is disordered.
Proteins fold via a balance of hydrophobic interactions (which need
bulk water as the ``away'' phase) and hydrogen bonds with the solvent.
No — anhydrous biomolecules are stable but inactive.
Q 9.29
What modification is done on the Ti plasmid of Agrobacterium
tumefaciens to convert it into a cloning vector?
Concept used. The Ti (tumour-inducing) plasmid of
Agrobacterium tumefaciens naturally transfers a segment called
T-DNA into the plant genome at the wound site, causing
crown-gall tumours. To use this plasmid as a cloning vector, the
tumour-inducing genes within the T-DNA region are removed (disarmed)
and replaced with the gene of interest plus a selectable marker. The
vir (virulence) genes that drive T-DNA transfer are kept intact, so
the plasmid still delivers the cargo to the plant.
The tumour-inducing genes of the T-DNA region are deleted and
replaced with the gene of interest; the disarmed Ti plasmid then carries
the gene into the plant genome via its retained vir system.
AS
Ankit Sharma
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Keep the delivery machinery, remove the disease.
Identify the two functional regions of Ti: T-DNA (cargo, includes
tumour genes) and vir region (transfer machinery).
Disarm the T-DNA: strip out the tumour genes, leave the border
sequences, insert the desired gene.
Reintroduce into Agrobacterium, then co-culture with plant
tissue. The bacterium does the rest.
Why this matters. This disarmed Ti plasmid is the basis of all
modern transgenic dicot crops (Bt cotton, Golden Rice, herbicide-tolerant
soybean).
Disarm the T-DNA (remove tumour genes), insert the gene of interest,
keep vir intact.
Short Answer Type Questions
Q 9.30
What is meant by gene cloning?
Concept used.Gene cloning is the production of many
genetically identical copies of a specific DNA segment (the gene of interest)
by:
0pt
Cutting the gene out of the source DNA with restriction enzymes;
Ligating it into a cloning vector that has its own
origin of replication;
Introducing the recombinant vector into a competent host cell by
transformation;
Letting the host divide so each daughter cell carries (and replicates)
a copy of the gene.
After overnight growth a single host cell yields millions of daughter cells,
each with many plasmid copies — a clone of the original gene.
State the goal: amplify a single gene to millions of copies in vivo.
Contrast with PCR (in vitro amplification): cloning produces a
living, expressible copy, while PCR produces a pure
molecular copy.
Gene cloning = producing many identical copies of a gene by
inserting it into a self-replicating vector and propagating the recombinant
vector in a host cell.
VK
Vivaan Kapoor
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. ``Cloning'' here means molecular cloning, not
organism cloning (e.g. Dolly the sheep). The product is many copies of a
single piece of DNA, not many identical organisms. Once that distinction is
clear, the workflow is just ``cut, paste, transfer, grow''.
The gene is excised from its native context by restriction-enzyme
digestion at flanking palindromic sites, producing a fragment with
defined sticky ends.
The same restriction enzyme linearises a cloning vector at its
multiple-cloning site, generating matched sticky ends.
DNA ligase joins fragment and vector via two new phosphodiester
bonds, producing a circular recombinant molecule.
The recombinant vector is introduced into a competent host
(E. coli via heat shock or electroporation) — this is the
transformation step.
As the host divides, the vector replicates from its ori
independently of the chromosome. After overnight growth, one cell
becomes ∼109 daughter cells, each carrying many copies
of the gene.
Recombinants are picked via the selectable marker (antibiotic or
blue-white screen) and used as a renewable source of the gene.
Why this matters. Gene cloning underlies recombinant-protein
production (insulin, hGH, vaccines), gene-function studies, the cataloguing
of genomes, and CRISPR-Cas9 guide design — modern biotech wouldn't exist
without it.
In-vivo amplification of a chosen DNA fragment by inserting it
into a self-replicating vector and propagating the recombinant vector in
a competent host cell.
Q 9.31
Both a wine maker and a molecular biologist who had developed a
recombinant vaccine claim to be biotechnologists. Who in your opinion is
correct?
Concept used.Biotechnology is defined by the
European Federation of Biotechnology (EFB) as ``the integration of natural
sciences and organisms, cells, parts thereof, and molecular analogues for
products and services''. By this broad definition, any use of living
cells to make a product is biotechnology. Wine fermentation by
Saccharomyces cerevisiae (a centuries-old traditional process) and
recombinant Hepatitis B vaccine production (a modern molecular process)
both qualify.
Apply the EFB definition: wine making uses live yeast to convert
sugars to ethanol + CO2 — biotechnology.
Vaccine making uses recombinant yeast expressing HBsAg — biotechnology.
Therefore both claims are correct. Traditional biotechnology and
modern (rDNA-based) biotechnology are both subsets of the discipline.
Both are correct. Traditional fermentation (wine) and modern
recombinant production (vaccine) are equally biotechnology.
SV
Sanya Verma
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The conflict here is one of vocabulary, not biology.
Once the EFB definition is applied, the dispute evaporates.
Map each practice to ``cell as factory'': wine = yeast as factory
for ethanol; recombinant vaccine = yeast as factory for HBsAg.
Both fit the definition; the only difference is the sophistication of
the cell engineering.
Why this matters. Biotech is a continuum from bread, beer, dahi
(traditional) to insulin and CAR-T cells (modern). NEET often tests this
breadth.
Both are biotechnologists.
Q 9.32
A recombinant DNA molecule was created by ligating a gene to a
plasmid vector. By mistake, an exonuclease was added to the tube containing
the recombinant DNA. How does this affect the next step in the experiment
i.e. bacterial transformation?
Concept used. An exonuclease chews nucleotides off the
ends of any linear DNA exposed in the tube. A correctly ligated recombinant
plasmid is circular (no free ends), so an exonuclease shouldn't touch
it. But the tube usually contains a mixture of:
Un-ligated linear vector and insert fragments (vulnerable).
Nicked or partially ligated species (vulnerable).
Exonuclease degrades all linear DNA in the tube; only intact circles
survive.
If ligation was efficient, intact recombinant circles still
transform, but at lower yield (because their preparation has been
net-degraded).
If ligation was incomplete, the recombinant fraction is mostly
eaten and transformation efficiency crashes; few or no recombinant
colonies appear.
Linear DNA is degraded; circular recombinant molecules survive but
yield is much reduced, so transformation efficiency falls sharply (and may
fail outright if ligation was incomplete).
PJ
Pranav Joshi
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The shape of the DNA decides its fate inside the
tube. Closed circular molecules have no free ends and survive an exonuclease;
linear molecules have two free ends and are eaten. So the question reduces to
``what fraction of the ligation product is genuinely circular?''.
Sort the tube contents by topology: (i) closed circular recombinant
(the ligation success product, fully resistant), (ii) nicked
circular (one strand still has a gap, somewhat vulnerable),
(iii) linear unligated vector and insert (fully vulnerable).
Add exonuclease: linear DNA is degraded to dNMPs within minutes;
nicked circles are slowly chewed from the nick; closed circles
remain intact throughout.
Outcome at the transformation step depends entirely on the surviving
circular recombinant fraction, which is usually only ∼ 5–20 %
of the total ligation mix. So if the lab considered transformation
efficiency before, it will be substantially lower now.
Recovery option: re-extract the surviving plasmid, re-quantify, and
try transformation again. The recombinant cells that do appear are
still genuine recombinants — just rarer.
Why this matters. The same shape-decides-fate logic is exploited
deliberately in the plasmid-safe DNase protocol, where exonuclease
is added after ligation to clean up linear contaminants without
touching the recombinant circles.
Reduces transformation efficiency by degrading linear DNA in the
tube; recombinant circles survive but the surviving pool is much smaller, so
fewer recombinant colonies are obtained.
Q 9.33
Restriction enzymes that are used in the construction of recombinant
DNA are endonucleases which cut the DNA at specific-recognition
sequence. What would be the disadvantage if they do not cut the DNA at
specific-recognition sequence?
Concept used. The whole cut-and-paste logic of rDNA
relies on predictability: an enzyme that recognises a specific
palindrome (e.g. 5'-GAATTC-3' for EcoRI) cuts every occurrence of that
palindrome and only those. This generates uniform, predictable sticky ends.
If restriction enzymes cut at random:
Fragments would have random end sequences and random lengths →
no way to predict where the cut falls in either vector or insert.
Sticky ends from the two halves of the same cut would no longer be
guaranteed to match the ends from a different cut on a different
molecule → no ligation.
The vector might be cut inside the ori or selectable marker,
ruining the cloning vehicle.
Even the cut sites cannot be reproduced from one experiment to the
next, so cloning becomes a one-off lottery rather than a routine
procedure.
Without sequence specificity, cuts would be random — vector and
insert would have unpredictable, mismatched ends; ligation would fail; the
ori/marker could be destroyed; the cut would be irreproducible.
Cloning would be impossible.
AK
Aarav Kumar
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. Specificity = predictability = reproducibility.
Lose the first and the other two vanish.
Without specificity, the location of each cut is a roll of dice.
Cut site might land inside ori, marker, gene of interest, or
anywhere else.
Sticky ends would be random, so the chance of two ends being
complementary and ligatable would drop to near zero.
Two separate cloning attempts would produce different fragment sets
from the same starting DNA — irreproducible.
Why this matters. The same logic explains why CRISPR-Cas9, which is
guided by a specific 20-nt sequence, is so much more useful than
random mutagenesis.
Random cuts = random ends = no ligation = no cloning.
Q 9.34
A plasmid DNA and a linear DNA (both are of the same size) have one
site for a restriction endonuclease. When cut and separated on agarose gel
electrophoresis, plasmid shows one DNA band while linear DNA shows two
fragments. Explain.
Concept used. A circular plasmid cut at a single site is
converted into one linear molecule of the same total length —
geometry-wise, one cut on a closed loop produces one line. A linear molecule
cut at a single site, however, is split into two pieces, one on either side
of the cut. The gel separates fragments by size, so:
0pt
Plasmid → one band (full length).
Linear DNA → two bands (one for each side of the cut).
Visualise the circular plasmid as a rubber band: a single cut snips
it into one piece (a line).
Visualise the linear DNA as a stretched string: a single cut splits
it at one point into two pieces.
The gel therefore shows one band for the plasmid and two bands for
the linear DNA.
Topology: one cut on a circle gives one linear piece (one band);
one cut on a line gives two pieces (two bands).
KB
Krishna Banerjee
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The difference is topological. Cutting a loop once
= a line. Cutting a line once = two lines.
Count the pieces directly: n cuts on a circle →n linear
fragments. n cuts on a line →n+1 linear fragments.
For n=1: circle → 1 piece, line → 2 pieces.
Gel shows correspondingly: plasmid 1 band, linear DNA 2 bands.
The plasmid's loop topology means one cut = one piece; the linear
DNA's open topology means one cut = two pieces.
Q 9.35
How does one visualise DNA on an agarose gel?
Concept used. DNA is colourless and invisible under white light. To
see it on an agarose gel:
0pt
Stain the gel (during or after the run) with
ethidium bromide (EtBr). EtBr intercalates between adjacent
base pairs of the DNA.
Illuminate the gel with UV light (∼ 300 nm) on a
UV transilluminator.
EtBr fluoresces orange (∼ 605 nm) at the DNA bands;
DNA-free regions stay dark.
Photograph through a UV-blocking filter to record the band pattern.
Stain with ethidium bromide, then expose to UV light on a
transilluminator — DNA bands fluoresce bright orange against a dark
background.
TK
Tara Kapoor
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Make DNA fluorescent, then shine the right
wavelength on it.
Soak the gel in EtBr solution (typically 0.5 μg/mL) for
∼ 10 min. EtBr inserts (intercalates) between base pairs.
Place the gel on a UV transilluminator. The intercalated EtBr absorbs
UV and re-emits visible orange light at ∼ 605 nm.
Capture the image with a CCD camera through a UV filter.
Why this matters. Newer dyes (SYBR Green, GelRed) are less mutagenic
than EtBr but rely on the same intercalation + fluorescence principle.
EtBr staining followed by UV transillumination.
Q 9.36
A plasmid without a selectable marker was chosen as vector for
cloning a gene. How does this affect the experiment?
Concept used. A selectable marker (e.g. antibiotic
resistance gene like ampR) lets us distinguish host cells that
have taken up the vector from those that have not. Without a marker, we have
no way to identify the rare cells that were successfully transformed.
After plating the transformation mixture, every colony will grow — both the
transformed and the non-transformed — but we cannot tell which are which.
Transformation is inherently inefficient: typically only 1 in
104–106 cells takes up a plasmid.
Without a marker, the transformed cells are mixed in among
∼105× more non-transformed cells.
Selecting recombinants becomes impossible because no
difference-of-growth is available.
No way to distinguish transformed from non-transformed cells; the
rare transformants are lost in the much larger population of untransformed
cells, so the cloning effectively fails at the selection step.
AS
Aditi Singh
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. A cloning experiment needs a filter between
``the few cells that worked'' and ``the many cells that didn't''. Marker is
that filter.
Without antibiotic-based selection, plating the mix on a normal
medium yields a confluent lawn of non-recombinants.
Recombinant colonies cannot be picked because they are
indistinguishable.
Why this matters. The very design of every commercial vector
(pUC, pBR322, pET) starts with a marker — it's the only practical way to
recover the rare transformants.
The experiment fails at selection: cannot identify transformed
cells.
Q 9.37
A mixture of fragmented DNA was electrophoresed in an agarose gel.
After staining the gel with ethidium bromide, no DNA bands were observed.
What could be the reason?
Concept used. For DNA to appear as a band on an EtBr-stained gel,
several conditions must be met: (i) the DNA must be present at high enough
concentration to be detected by EtBr fluorescence, (ii) it must be intact,
not degraded into nucleotides, (iii) the EtBr must intercalate into it, and
(iv) the gel must be viewed under UV light.
DNA concentration too low — below the EtBr detection limit
(∼ 1–10 ng per band).
DNA fully degraded — small mono/oligonucleotides run off the gel and
give no band.
Gel illuminated under visible light rather than UV — EtBr fluorescence
only shows under UV.
EtBr staining step was skipped, ineffective, or used at too low a
concentration to intercalate.
Most likely causes: (i) too little DNA, (ii) DNA fully degraded,
(iii) UV transilluminator not used, or (iv) faulty/missed EtBr staining.
DM
Diya Mehta
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Walk the path from sample to image and look for the
break.
Sample side: was there enough DNA loaded? Was it degraded before
loading?
Detection side: did the EtBr actually stain? Did we expose to UV
(not white light) for visualisation?
Why this matters. Troubleshooting a missing gel band is part of
weekly lab life. Knowing the four checkpoints — concentration, integrity,
stain, light source — saves days.
Insufficient DNA, degraded DNA, no UV illumination, or missed/failed
EtBr staining.
Q 9.38
Describe the role of CaCl2 in the preparation of competent
cells?
Concept used. The bacterial cell wall is negatively charged
(phosphates on the lipopolysaccharide), and so is DNA (phosphates on the
backbone). Two negatives repel — so DNA cannot approach the cell. Divalent
Ca^2+ ions neutralise both surfaces, screening the repulsion
and letting DNA stick to the cell envelope. A subsequent heat shock at
42 ∘C creates transient pores through which the bound DNA enters.
Soak cells in ice-cold CaCl2 (0.1 M typical). Ca^2+
binds to the cell wall and to phosphate groups on the DNA.
Mix cells with the recombinant DNA, keep on ice — DNA-cell
complexes form on the wall.
Heat shock (42 ∘C, 90 s) opens transient pores; DNA enters
the cell.
Cool back, recover in rich medium, then plate on selection.
CaCl2 provides Ca^2+ ions that neutralise the negative
charges on the cell wall and on DNA, letting DNA bind to the cell envelope
before the heat-shock pores let it in.
YI
Yash Iyer
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Ca^2+ solves an electrostatic
problem; heat shock solves a permeability problem. Both are needed.
Treat the wall and DNA as parallel-plate capacitors with negative
charge densities. Ca^2+ ions act as counter-ions that flatten
the potential between them.
Once the charge barrier is gone, DNA adsorbs onto the cell surface
and is poised to be pulled inside by the heat-shock pulse.
Why this matters. Without CaCl2 the transformation efficiency
drops by 3–4 orders of magnitude. The simple chemistry of charge
neutralisation drives the entire cloning workflow.
Neutralises negative charges on both DNA and cell wall, letting
DNA bind the cell surface ahead of heat-shock-induced uptake.
Q 9.39
What would happen when one grows a recombinant bacterium in a
bioreactor but forget to add antibiotic to the medium in which the
recombinant is growing?
Concept used. The recombinant plasmid carries an antibiotic-resistance
marker. Carrying the plasmid is a metabolic burden on the cell —
replication, transcription and translation of the plasmid and its gene cost
energy. In the presence of the antibiotic, only resistant (plasmid-bearing)
cells survive, so the population stays 100 % recombinant.
Without the antibiotic the selection pressure vanishes:
Cells that spontaneously lose the plasmid (a rare event per
division) escape the metabolic cost and grow slightly faster than
plasmid-bearing cells.
Over many generations, plasmid-free cells out-compete the
recombinant cells and gradually dominate the bioreactor.
Yield of the recombinant protein drops sharply as the recombinant
fraction shrinks.
Even worse, the dominant non-recombinant population will continue
to consume nutrients without producing the target protein.
Plasmid-free cells (faster-growing because they avoid the metabolic
burden) will out-compete recombinant cells over generations; the bioreactor
soon produces little to no recombinant protein.
MS
Meera Sharma
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle. The antibiotic is the population-level filter that
keeps the recombinants on top. Remove the filter and the faster-growing
non-recombinants win.
Population dynamics: recombinant cells reproduce slightly slower
because of plasmid burden. Without antibiotic, faster non-recombinant
cells take over.
Result: yield drops, the bioreactor becomes economically useless.
Why this matters. This is also why ``plasmid loss'' is one of the
top failure modes in industrial recombinant protein production — and why
genome-integrated expression is sometimes preferred for long runs.
Loss of selection pressure ⇒ plasmid-free cells
out-compete recombinant cells ⇒ recombinant protein yield drops
sharply.
Q 9.40
Identify and explain steps `A', `B' and `C' in the PCR diagram given below.
Fig. 11.1, NCERT Exemplar Class 12 Biology, Chapter 11 (Biotechnology: Principles and Processes).
Concept used.Polymerase Chain Reaction (PCR) amplifies a
specific DNA segment through repeated cycles of three temperature-defined
steps. The figure labels the three steps as A, B and C:
0pt
A — Denaturation. The double-stranded template DNA is heated
to ∼ 94–95 ∘C, which breaks the hydrogen bonds
between the two strands. The result is two single strands. Arrow
``Heat'' in the figure.
B — Annealing. Temperature is dropped to ∼ 50–60 ∘C
so that the two short primers (one for each strand) can
base-pair with their complementary sequences flanking the region to
be amplified.
C — Extension (Elongation). Temperature is raised to
∼ 72 ∘C, the optimum for Taq DNA polymerase.
Taq adds deoxynucleotides to the 3'-OH end of each annealed primer,
synthesising a new strand complementary to the template.
A (Denaturation): heat at 94–95 ∘C separates the two
parental strands.
B (Annealing): cool to 50–60 ∘C, primers bind the
single-stranded templates at their target sequences.
C (Extension): warm to 72 ∘C, Taq polymerase extends each
primer into a full complementary strand using dNTPs.
Repeat A–B–C for ∼ 30 cycles. After n cycles, the number
of double-stranded copies of the target region is
N = N0 × 2n, i.e. ∼109-fold amplification.
A = Denaturation; B = Annealing of primers; C = Extension by
Taq DNA polymerase.
IR
Ishita Reddy
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Picture-first. Read the figure top-to-bottom. The ds-DNA at the top
is heated (A) into two strands, primers stick on them (B), and the Taq
polymerase fills in the gap (C). After 30 cycles (∼ 1 billion times)
the dotted region is amplified.
A — denaturation at 94–95 ∘C, breaks H-bonds, strands
separate. ``Heat'' arrow in figure.
B — annealing at 50–60 ∘C, primers (small grey blocks
with arrows) bind the flanking sequences.
C — extension at 72 ∘C, Taq polymerase elongates each
primer with dNTPs, producing two new ds-DNAs.
Cycle repeats; after 30 cycles, ∼109 identical copies
of the target region exist in the tube.
Why this matters. PCR is the most widely used DNA-amplification
technique in the world — COVID RT-PCR tests, forensic DNA fingerprinting,
prenatal diagnosis, and almost all modern cloning experiments rely on it.
A = denaturation, B = primer annealing, C = extension by
Taq DNA polymerase.
Concept used. The figure shows pBR322, the classic
E. coli cloning vector developed by Bolivar and Rodriguez in 1977.
The plasmid is 4361 bp long and carries (i) an origin of replication
(ori), (ii) two antibiotic-resistance genes (ampR for
ampicillin and tetR for tetracycline), and (iii) several unique
restriction sites suitable for cloning (EcoRI, ClaI, HindIII, BamHI, SalI,
PvuI, PvuII). Reading the labelled and unlabelled regions:
0pt
A — ampR: the ampicillin-resistance gene
(encodes β-lactamase). Cells carrying intact ampR
survive on ampicillin plates.
B — BamH I (BamHI) restriction site: a unique
cloning site inside tetR. Insertion at this site
inactivates tetracycline resistance, providing the basis for
insertional-inactivation screening of recombinants.
C — tetR: the tetracycline-resistance gene.
Intact tetR confers tetracycline resistance.
Recall the standard pBR322 map: two antibiotic markers
ampR (1 o'clock–3 o'clock arc) and tetR
(9 o'clock–12 o'clock arc), plus ori (5 o'clock) and
rop (6 o'clock).
Map labels to the figure: A on the right belongs to the
ampicillin-resistance arc; C on the upper-left belongs to the
tet-resistance arc; B on the left is the BamHI restriction site
(inside tetR).
A =ampR (ampicillin-resistance gene);
B = BamHI restriction site (inside tetR);
C =tetR (tetracycline-resistance gene).
RP
Rohit Pillai
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. Decode the map by combining the labelled landmarks
(EcoRI, ClaI, HindIII, SalI, PvuI, PvuII, ori, rop) and the
positions of A, B and C.
The right-side arc (between HindIII at top and SalI at right) is the
classic tetR arc — so the unlabelled region in this arc,
labelled tetR in the figure, matches tetR.
Region A on the right is ampR.
The unlabelled cut just left of PvuI on the upper left is the
BamHI restriction site — region B.
The label C sits in the upper-left arc, marking the
tetR gene (the tetracycline-resistance arc).
Why this matters. pBR322 is the ``Hello World'' vector of molecular
biology. Knowing its map by heart is a core CBSE/NEET expectation.
A =ampR; B = BamHI site;
C =tetR.
Long Answer Type Questions
Q 9.42
For selection of recombinants, insertional inactivation of
antibiotic marker has been superceded by insertional inactivation of a marker
gene coding for a chromogenic substrate. Give reasons.
Concept used. In the classical insertional-inactivation
scheme using two antibiotic markers (e.g. ampR and
tetR of pBR322), recombinant selection takes two steps:
(i) grow all transformants on ampicillin plates, and (ii) replica-plate
onto tetracycline plates to identify the tetS (recombinant)
colonies. This requires duplicate plates and the labour of replica-plating.
The newer blue-white screening replaces one antibiotic marker
with a marker gene encoding a chromogenic enzyme — most commonly the
lacZα fragment of β-galactosidase. The vector carries
a multiple cloning site (MCS) inserted in the middle of lacZα.
When plated on a medium containing X-gal (a colourless substrate)
and the inducer IPTG:
0pt
Non-recombinant colonies have intact lacZα, make active
β-galactosidase, cleave X-gal to a blue product, and form
blue colonies.
Recombinant colonies have a foreign DNA insert disrupting
lacZα, cannot cleave X-gal, and form
white colonies.
So we identify recombinants on a single plate in a single growth step —
much faster and cheaper than two-antibiotic replica plating.
Compare workflow lengths: two-marker antibiotic screening requires
primary plate + replica plate + comparison. Blue-white screening
needs only one plate.
Compare resource use: two-antibiotic method needs two antibiotics
and replica-plating apparatus. Blue-white needs only one antibiotic
plus X-gal/IPTG.
Compare ease of scoring: tet-sensitivity needs colony-by-colony
comparison between two plates. Blue/white is a direct visual readout
on one plate.
Compare safety: X-gal is non-toxic to the user. The duplicate
antibiotics aren't hazardous either, but the overall reagent burden
is lower with blue-white.
Practical bonus: blue-white plates can be photographed and counted
automatically; replica plates need manual eye-balling.
Blue-white screening using lacZα insertional
inactivation needs only one plate (vs two with the antibiotic-only
method), gives a direct colour readout (vs growth-vs-no-growth
comparison), avoids replica-plating labour, and uses fewer reagents — so it
has replaced the older antibiotic-only insertional inactivation.
VS
Vivaan Sharma
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. The comparison is essentially time, labour and
clarity. List those three axes and the new method wins on all of them.
Time: classical method needs overnight growth on plate 1, then
overnight growth on plate 2 after replica-plating (∼ 36–48 h).
Blue-white needs only overnight growth on plate 1 (∼ 16–24 h).
Half the time.
Labour: replica-plating each colony from plate 1 to plate 2 is
manual and error-prone. Picking white colonies from a single X-gal
plate is direct.
Clarity of readout: with two antibiotics you must compare presence
on plate 1 with absence on plate 2 — easy to mis-score colonies that
grew weakly. With blue-white, white = recombinant, blue = empty
vector. Binary, unambiguous.
Scalability: blue-white is amenable to high-throughput colony pickers
(which detect colour). Replica-plate selection is not.
Cost: one antibiotic + X-gal + IPTG is cheaper than two antibiotics
+ replica-plating consumables for an industrial pipeline.
Why this matters. The same logic — replace ``growth vs no-growth''
selection with a direct readout — drives modern fluorescent-protein
co-expression and FACS-based selection. Visual signals scale better than
viability signals.
Blue-white screening (single plate, direct colour readout, no
replica-plating) is faster, simpler, cheaper, and easier to score than
two-antibiotic insertional inactivation; hence it has superseded the older
method.
Q 9.43
Describe the role of Agrobacterium tumefaciens in
transforming a plant cell.
Concept used.Agrobacterium tumefaciens is a soil
bacterium that naturally infects wounded dicot plants and causes
crown-gall tumours. The infection is mediated by a large
plasmid called the Ti (Tumour-inducing) plasmid. Two regions of
Ti are critical:
0pt
T-DNA (transferred DNA) — a 20–25 kb segment delimited
by two ``border'' sequences. T-DNA is excised from the Ti plasmid,
transferred into the plant cell, and integrated into the
plant chromosomal DNA.
Virulence (vir) region — a cluster of genes whose
products process and shuttle T-DNA into the plant.
Step-wise natural infection.
A wounded plant releases phenolic compounds (e.g. acetosyringone)
that activate the vir genes in nearby Agrobacterium.
Agrobacterium attaches to the plant cell wall using surface
proteins.
vir gene products nick the T-DNA borders, excise the T-DNA as
a single-stranded molecule (T-strand), and coat it with VirD2 and
VirE2 proteins for protection.
A bacterial Type IV secretion system ferries the
T-DNA-protein complex into the plant cell.
Inside the plant cell, nuclear localisation signals on VirD2/VirE2
drive the T-DNA into the nucleus.
The T-DNA is integrated at random sites into the plant chromosomes
by the host's own DNA-repair machinery. Genes carried on the T-DNA
are now transcribed by plant RNA polymerase.
In nature, the T-DNA encodes auxin and cytokinin biosynthesis genes
(causing tumour formation) plus opine synthesis genes (which feed
Agrobacterium).
Engineering for plant transformation.
Engineers disarm the Ti plasmid by deleting the tumour-causing
auxin/cytokinin genes from the T-DNA.
The gene of interest plus a plant-selectable marker (e.g.
nptII for kanamycin resistance) is inserted between the
T-DNA borders.
The recombinant Ti is reintroduced into Agrobacterium, and
plant tissue (leaf disc, callus) is co-cultured with this strain.
Agrobacterium transfers the engineered T-DNA into the plant
genome via its retained vir machinery — exactly as it would
in nature.
Transformed plant cells are selected on kanamycin, regenerated into
whole plants, and screened for stable integration and expression of
the foreign gene.
Agrobacterium tumefaciens acts as a natural
plant-transformation vector: its Ti plasmid's T-DNA is excised and
transferred (via the vir-encoded type-IV secretion system) into the
plant cell nucleus, where it integrates into the chromosomes. Biotechnologists
disarm the T-DNA, replace the tumour genes with the gene of interest plus a
plant marker, and exploit Agrobacterium's natural delivery to
produce transgenic plants.
KM
Karan Mehta
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Strategic angle.Agrobacterium is nature's only known
gene-delivery system that transfers DNA across kingdoms (bacterium →
plant). Biotech keeps the machinery and replaces the cargo.
Native role: Agrobacterium infects wounded dicot plants
and induces tumours by transferring T-DNA (carrying auxin, cytokinin
and opine genes) into the plant genome.
Mechanism: phenolic wound signal → activates vir genes
→ nicks T-DNA borders → excises T-strand → shuttles it
through a type-IV secretion channel → T-DNA reaches plant
nucleus → random integration into chromosome → plant
expresses bacterial genes.
Engineering retains the vir machinery and the T-DNA borders
but replaces the cargo with the gene of interest plus a plant
marker. Result: a disarmed delivery system.
Applied protocol: co-culture leaf discs or callus with the
engineered Agrobacterium strain, select on antibiotic,
regenerate transgenic plants via tissue culture.
Quality control: confirm single-copy integration by Southern blot
or PCR; confirm expression by Northern blot / Western blot / direct
phenotype.
Real example: Bt cotton was made by transferring the cry1Ac
gene from Bacillus thuringiensis into cotton genome using
disarmed Ti. The resulting plants produce Bt toxin that kills
bollworms.
Why this matters. The same disarmed-Ti workflow underpins almost
every commercial dicot transgenic — a single engineered bacterium feeds a
multi-billion-dollar industry.
Agrobacterium tumefaciens uses its Ti plasmid to transfer
T-DNA into plant chromosomes via the vir-encoded type-IV secretion
system. Disarming the T-DNA and replacing tumour genes with the gene of
interest converts it into a universal plant-transformation vector.
Q 9.44
Illustrate the design of a bioreactor. Highlight the difference
between a flask in your laboratory and a bioreactor which allows cells to
grow in a continuous culture system.
Concept used. A bioreactor is a closed vessel designed to
grow microbial, plant or animal cells under tightly controlled physical and
chemical conditions so that they convert raw materials into a target
biological product. The most widely used industrial bioreactor is the
stirred-tank bioreactor (Fig. alongside).
Schematic design of a stirred-tank bioreactor.
!%
[See diagram in the PDF version]
Key components of a stirred-tank bioreactor.
0pt
Agitator (impeller). Driven by a motor; keeps the cells
suspended and the broth uniformly mixed.
Sparger (air inlet). Bubbles sterile air through the broth
from the bottom, supplying oxygen to aerobic cells.
Cooling jacket. Removes the heat released by cellular
metabolism to keep temperature constant (typically
∼ 30–37 ∘C).
Sensors and probes. Continuously monitor pH, dissolved
oxygen and temperature; feedback to controllers that adjust
acid/base or air flow.
Inlets for nutrients, acid/base, antifoam.
Outlets for harvest, sampling and exhaust gases.
Foam breaker at the top to prevent foam overflow.
Laboratory flask vs. continuous-culture bioreactor.
Scale-up suitability & Not scalable beyond a few litres & Designed for industrial-scale production
tabular
A lab flask is a closed batch vessel with no flow of medium and only
passive aeration. Cells grow to stationary phase in hours to days,
then stop producing.
A continuous-culture bioreactor is a controlled vessel with steady
flow of fresh medium in and used medium out. Cells stay in log phase
for days or weeks, producing the target protein continuously.
The bioreactor's combined control of temperature, pH, dissolved
O2 and agitation maximises growth rate and product yield.
A shake flask cannot do this.
A bioreactor is a sterile, agitated, aerated, sensor-controlled
vessel with inlets and outlets that lets cells grow continuously in log phase
at industrial scale; a lab flask is a small, batch-only, manually-shaken
vessel with no environmental control. The bioreactor gives orders-of-magnitude
higher and longer-running product yield.
TI
Tara Iyer
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle. A bioreactor is a flask plus control,
flow and scale. Walk those three axes.
Control. A flask sits on a shaker; pH, oxygen and
temperature drift uncontrolled. A bioreactor has sensors and feedback
loops that hold every parameter at its setpoint.
Flow. A flask is a closed batch — once nutrients are gone
and waste has built up, cells die. A continuous-culture bioreactor
constantly removes used medium and adds fresh nutrients, holding the
cells indefinitely in their most productive (log) phase.
Scale. A flask is litres at most. A bioreactor is hundreds
to tens of thousands of litres, with impeller-and-sparger geometries
that keep mixing and aeration uniform across the entire volume.
Aeration in detail. A flask depends on diffusion through a
cotton plug — fine for litres, hopeless for tonnes. A bioreactor
sparges sterile air from the bottom, while the impeller breaks the
bubbles into a fine dispersion for high oxygen transfer rates.
Sterility. A flask is autoclaved before inoculation and
otherwise left to its own devices. A bioreactor has sterile-filter
inlets/outlets, steam-in-place jackets, and antifoam pumps to keep
the culture contaminant-free for weeks of continuous operation.
Sampling and harvest. A flask is end-of-batch only. A
bioreactor has dedicated sampling and harvest ports so the operator
can monitor cell density, metabolite levels and product titre in
real time.
Net outcome. The bioreactor turns a fragile shake-flask
culture into a robust, predictable, high-yield industrial process.
Why this matters. Every commercial recombinant protein — insulin,
growth hormone, erythropoietin, monoclonal antibodies, COVID vaccines — comes
out of large continuous or fed-batch bioreactors. The flask is a teaching
tool; the bioreactor is the factory.
Bioreactor = scale + control + flow. A lab flask is small,
uncontrolled, batch-only; a continuous-culture bioreactor is large,
sensor-controlled, with steady medium inflow and product outflow, holding
cells in log phase for industrial-scale, high-yield production.
NCERT Exemplar Solutions for Class 12 Biology: All Chapters
Biotechnology Principles and Processes Class 12 Biology Exemplar Solutions FAQs
Ques. Where can I download Class 12 Biology Chapter 9 Biotechnology Principles and Processes Exemplar Solutions PDF?
Ans. The Biotechnology Principles and Processes Exemplar Solutions PDF is downloadable directly from this page. It is a 72-page document with worked solutions to all 38 Exemplar problems (12 MCQ, 5 MCQ-II, 9 VSA, 8 SA, 4 LA), aligned to the 2026-27 NCERT.
Ques. How many problems are in the Class 12 Biology Chapter 9 NCERT Exemplar?
Ans. The NCERT Exemplar for Biotechnology: Principles and Processes carries 38 problems split across five formats: 12 MCQ (single correct), 5 MCQ-II (multiple correct), 9 VSA, 8 SA and 4 LA.
Ques. Are these Exemplar Solutions aligned with the 2026-27 syllabus?
Ans. Yes. The 2026-27 NCERT kept Biotechnology Principles and Processes intact (no trims), so every Exemplar problem maps to a live syllabus topic. Each solution flags the NCERT section it corresponds to.
Ques. Why should NEET aspirants solve the Class 12 Biology Chapter 9 Exemplar?
Ans. NEET pulls 3 to 5 questions from this chapter every year, several as MCQ-II / assertion-reason items. The Exemplar drills the exact terminology (palindrome, sticky end, MCS, selectable marker, sparger, downstream processing) that NEET keys reward. Solving all 38 problems builds the recall scaffold for the chapter.
Ques. Are the Exemplar Solutions for restriction enzymes the same as NCERT chapter Solutions?
Ans. No. The NCERT chapter Solutions cover the 11 end-of-chapter questions. The Exemplar Solutions cover the additional 38 Exemplar problems, which are tougher, more NEET-aligned, and test multi-correct MCQ formats absent in the NCERT chapter.
Ques. What is the most-asked Exemplar topic from Biotechnology Principles and Processes?
Ans. Restriction enzyme nomenclature and cut-site identification (Exemplar MCQs 1 to 4 and SA-5) form the most-asked cluster, reused in NEET 2025, 2024 and 2023. The 9-step rDNA workflow LA-2 is the highest-yield CBSE long-answer.



































































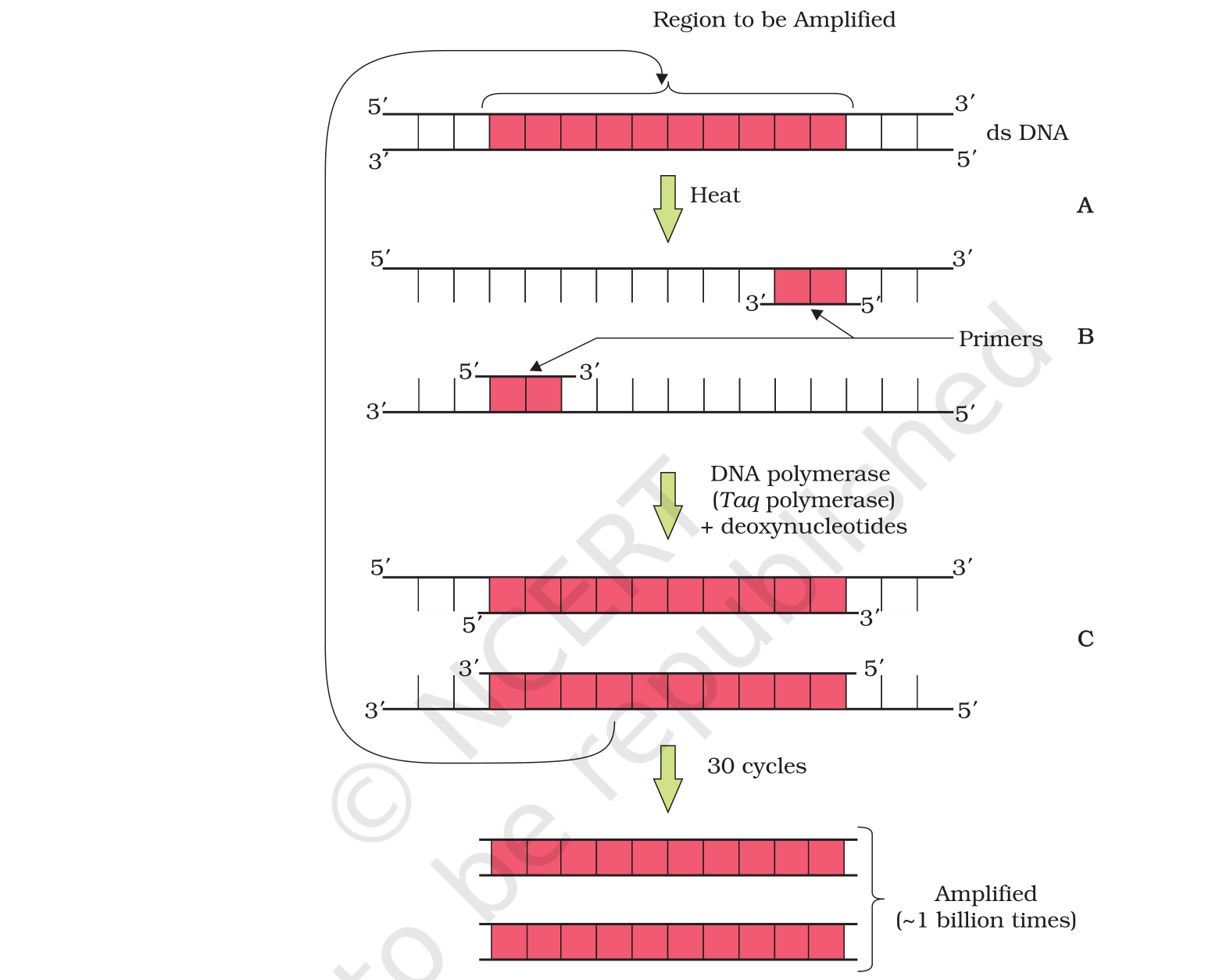
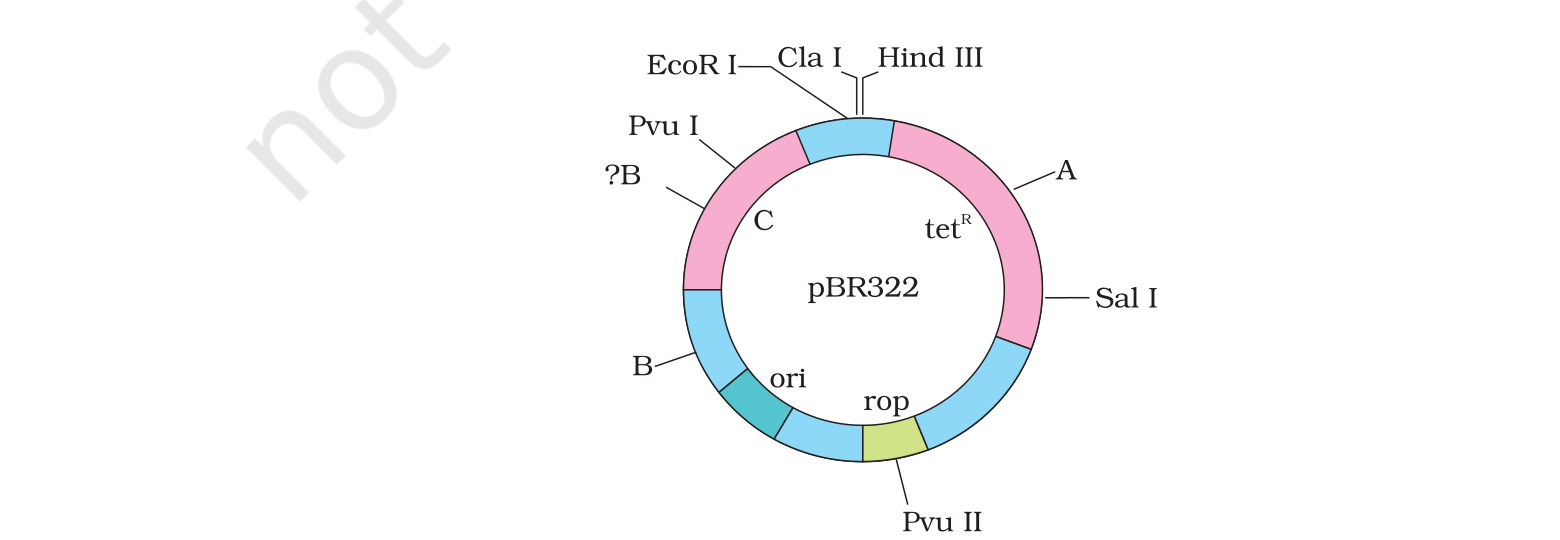


Comments