Senior Chemistry Editor | M.Sc. Chemistry, 12 Years | Updated on - May 25, 2026
The Alcohols, Phenols and Ethers chapter is the gateway to the entire Class 12 Chemistry organic block, carrying over 32 NCERT textbook exercise problems plus 12 intext questions spread across 26 pages of the current NCERT print. The solutions on this page walk you through every named reaction, distinction test, and mechanism question as per the 2026-27 syllabus.
CBSE Weightage: 6-8 marks (Unit 7 of the rationalised syllabus, combined with Haloalkanes block in some blueprint variants).
JEE Main Weightage: 3-5% of the Chemistry section, usually 1-2 questions on acidity order or named reactions.
NEET Weightage: 2-3 questions, with phenol reactions (Kolbe, Reimer-Tiemann) being recurring favourites.
Chapter 7 Alcohols, Phenols and Ethers NCERT Solutions PDF
What's inside this PDF: all 32 exercise questions + 12 intext questions solved with full mechanism arrows for SN1, SN2, dehydration, and electrophilic aromatic substitution; comparative acidity tables for ortho/meta/para substituted phenols; and a one-page named-reactions cheat sheet at the back.
Collegedunia's solution set is reviewed by chemistry educators with NEET-PG mentoring experience, so each step explicitly states which reagent attacks which centre, why the regiochemistry is the way it is, and what trap CBSE typically sets in the 3-mark variant of the same question.
How will Collegedunia's NCERT Solutions Help You Score in Class 12 Chemistry?
Organic chemistry rewards the student who can write a mechanism, not just a product. The solutions on this page give you the arrow-pushing diagram, the intermediate, and the by-product for every reaction that NCERT mentions, including the ones that the textbook itself only sketches in two lines.
Three habits this resource will build:
Distinction test fluency: Lucas test, ceric ammonium nitrate test, Victor-Meyer test, ferric chloride test for phenols. Every distinction-type question in the exercise is solved with the colour change written out.
Reagent recall: the solutions colour-code reagents by role (oxidising / reducing / electrophile-generator) so you stop confusing PCC with KMnO4 the night before the board.
IUPAC naming for poly-functional compounds with two or more substituents on the ring, which is one of the most-asked 2-mark questions in CBSE 2025.
NCERT Class 12 Chemistry Chapter 7 Alcohols, Phenols and Ethers Exercise-wise Question Map
The chapter splits its 32 main-exercise questions into nomenclature, preparation, reactions, and mechanism categories. The table below tells you which exercise number maps to which sub-topic so you can revise targeted clusters instead of front-to-back.
Exercise Range
Sub-topic
Question Count
Difficulty
7.1 - 7.4
Classification & IUPAC nomenclature
4
Easy
7.5 - 7.10
Preparation of alcohols (hydration, hydroboration, Grignard)
6
Medium
7.11 - 7.16
Reactions of alcohols (oxidation, dehydration, esterification)
6
Medium
7.17 - 7.22
Phenol preparation & acidity
6
Medium
7.23 - 7.28
Electrophilic substitution on phenol (Kolbe, Reimer-Tiemann)
6
Hard
7.29 - 7.32
Ethers - Williamson synthesis, cleavage
4
Medium
Use the cluster mapping to schedule three 45-minute revision sittings: nomenclature + ethers in one, alcohol reactions in another, and phenol reactions in the final pass.
Alcohols, Phenols and Ethers Class 12 Chemistry Important Named Reactions
Six named reactions dominate the board and entrance papers for this chapter. The solutions PDF walks through each one with the reagent set, conditions, and product. A compact recall list:
Named Reaction
Starting Material
Reagent / Conditions
Product Type
Kolbe's reaction
Phenol (sodium phenoxide)
CO2, 400 K, 4-7 atm; then H+
Salicylic acid
Reimer-Tiemann reaction
Phenol
CHCl3 + NaOH, then H3O+
Salicylaldehyde
Williamson synthesis
Alkyl halide + sodium alkoxide
Dry conditions, SN2
Ether
Hydroboration-oxidation
Alkene
B2H6 / THF, then H2O2 / OH-
Anti-Markovnikov alcohol
Lucas test
1degree / 2degree / 3degree alcohol
Conc. HCl + ZnCl2
Turbidity timing distinguishes class
Victor-Meyer test
1degree / 2degree / 3degree alcohol
P/I2, AgNO2, HNO2, NaOH
Red / blue / no colour
Every named reaction listed above has appeared at least once between CBSE 2021 and CBSE 2025, so memorising the reagent column alone is worth roughly 4 marks.
Alcohols, Phenols and Ethers Mechanism Walkthroughs in the Solutions PDF
NCERT explicitly asks for the mechanism in three places in the chapter exercise (acid-catalysed dehydration, acid-catalysed hydration, and Williamson synthesis). The PDF draws the curved-arrow steps for each, including the carbocation intermediate, the loss of water, and the deprotonation step.
Mechanism question tip: always label the rate-determining step. CBSE has docked 1 mark in 3-mark mechanism questions for unlabelled RDS in 2024 and 2025. The Collegedunia solutions box the RDS arrow in red so you don't forget it during revision.
Acidity Order of Phenols: The Single Highest-Yield Concept of Chapter 7
Whether the exam is CBSE, JEE Main, or NEET, a substituted-phenol acidity ranking question appears almost every year. The trick is to remember electron-withdrawing groups at ortho / para stabilise the phenoxide, while electron-donating groups destabilise it. Meta-position EWGs only contribute through inductive effect, not resonance.
Compound
pKa
Relative Acidity
2,4,6-trinitrophenol (picric acid)
0.4
Highest
p-nitrophenol
7.1
High
o-nitrophenol
7.2
High
Phenol
10.0
Reference
p-cresol (4-methylphenol)
10.2
Lower
p-methoxyphenol
10.2
Lower
The solutions PDF includes a side-bar derivation showing the four resonance structures of p-nitrophenoxide, which is the diagram most candidates draw incorrectly in board exams.
NCERT Class 12th Chemistry Chapter 7 Previous Year Question Trend
Below is a five-year scan of how Chapter 7 questions appeared across CBSE Boards, JEE Main, and NEET. The exam-comparison ordering leads with the latest held edition.
Year
CBSE Board
JEE Main
NEET
2026
Pending (May 2026 sitting)
1 question (Jan session) on Williamson synthesis
Pending (exam rescheduled)
2025
3-mark: distinguish 1degree, 2degree, 3degree alcohols by Lucas test
2 questions on phenol acidity, ether cleavage
2 questions on Kolbe and Reimer-Tiemann
2024
5-mark: phenol preparation from cumene + 2 reactions
1 question on hydroboration-oxidation
1 question on acidity of substituted phenols
2023
2-mark: IUPAC name of 2,4-dichlorophenoxyacetic acid
2 questions on Williamson + dehydration mechanism
2 questions on Lucas test + ether nomenclature
2022
3-mark: mechanism of acid-catalysed dehydration of ethanol
1 question on bond angles in ether
1 question on phenol oxidation
2021
3-mark: explain why phenol is more acidic than ethanol
-
1 question on alcohol classification
The pattern is clear: phenol acidity, named reactions, and Lucas test recur in some form every year. A student who masters these three sub-topics is statistically guaranteed 5-7 marks from Chapter 7 alone.
Common Mistakes Students Make in Alcohols, Phenols and Ethers
Top three mistakes flagged in CBSE evaluator notes (2024-2025):
Writing Markovnikov product in the hydroboration-oxidation step. Hydroboration is strictly anti-Markovnikov.
Confusing the order of Kolbe's reaction: students write the carboxylation step before forming the phenoxide. The correct sequence is phenol -> NaOH -> sodium phenoxide -> CO2 -> salicylate -> H+ -> salicylic acid.
Forgetting to mention dry ether as solvent in Williamson synthesis. Wet conditions hydrolyse the alkoxide nucleophile, and CBSE docks 0.5 marks for this omission.
Alcohols Phenols and Ethers Quick Formula and Concept Recall
Five high-recall facts to revise the night before any chapter test:
Boiling point order for isomers: 1degree > 2degree > 3degree alcohol (due to less steric hindrance to H-bonding).
Reactivity of alcohols with HX: 3degree > 2degree > 1degree (SN1 stability).
In phenol, the C-O bond is shorter than in alcohols because of partial double-bond character from ring conjugation.
Ether cleavage with HI gives alkyl iodide of the smaller group + alcohol of the larger group at lower temperature; at higher temperature, both products are alkyl iodides.
Preparation of Alcohols from Aldehydes, Ketones, and Carbonyl Compounds: Reduction and Grignard Routes
Two preparation families dominate NCERT exercise Q 7.5 to Q 7.10. The solutions PDF works each one with the reagent set and the alcohol class produced.
Reduction of aldehydes and ketones: R-CHO + 2[H] -> R-CH2-OH (1 degree alcohol) using NaBH4 or LiAlH4; R2C=O + 2[H] -> R2CH-OH (2 degree alcohol). Catalytic hydrogenation with Ni / Pt / Pd works for both, but reduces C=C alongside C=O.
Grignard alcohol synthesis: R-MgX adds to a carbonyl in dry ether, then aqueous work-up hydrolyses the magnesium alkoxide to the alcohol. The carbonyl picked controls the alcohol class: HCHO gives 1 degree, RCHO gives 2 degree, R2CO gives 3 degree. The PDF solves a Grignard preparation of 2-methylbutan-2-ol from acetone and ethyl magnesium bromide step by step.
Hydroboration-oxidation: alkene + B2H6 / THF, then alkaline H2O2 gives the anti-Markovnikov alcohol with syn-addition stereochemistry. The boron attaches to the less-substituted carbon; oxidation replaces it with -OH without rearrangement.
Acid-catalysed hydration: alkene + H2O / dil. H2SO4 gives the Markovnikov alcohol via a carbocation intermediate. Rearrangement is possible; the PDF flags the carbocation rearrangement trap on Q 7.7.
Phenol Preparation: Cumene Process, Dow Process, and Sodium Phenoxide Routes
NCERT lists four industrial and laboratory preparation routes to phenol. The solutions PDF works each with stoichiometry, conditions, and a one-line why-it-works gloss.
Route
Starting material
Reagent / conditions
Why this route
Cumene process
Cumene (isopropylbenzene)
Air (O2), then dil. H2SO4
Industrial route; acetone is the valuable co-product
Dow process
Chlorobenzene
NaOH, 623 K, 320 atm; then H+
Older industrial route; requires extreme T, P
Sulphonic acid fusion
Benzene sulphonic acid
Molten NaOH; then H+
Lab scale; goes via sodium phenoxide
Diazonium hydrolysis
Benzene diazonium chloride
Warm water
Best lab route from aniline
The cumene process is the most-asked preparation question (CBSE 2024, JEE Main 2023 January shift). Always mark acetone as a by-product, since CBSE allots 1 mark for naming the co-product.
NCERT exercise asks for at least three distinguishing-test answers. The solutions PDF tabulates the reagent, the colour change, and the canonical CBSE phrasing for each.
Lucas test: Lucas reagent is conc. HCl + anhydrous ZnCl2. Tertiary alcohols give immediate turbidity (carbocation forms instantly), secondary alcohols give turbidity in 5 to 10 minutes, primary alcohols give no turbidity at room temperature. The Lucas test class 12 distinction is asked in CBSE 2025 as a 3-mark question.
Iodoform test: I2 + NaOH on a methyl-carbinol (CH3-CHOH-R) or ethanol gives a yellow precipitate of CHI3. Methanol and 3 degree carbinols without the CH3-CHOH- pattern do NOT respond.
Ferric chloride test for phenol: phenol + neutral FeCl3 gives a violet colouration; alcohols do not respond. The Collegedunia solutions PDF reproduces the colour-change strip for revision.
Phenol Acidity vs Alcohol: Substituent Effects and Picric Acid Preparation
Phenol acidity vs alcohol is the highest-yield comparison question across the chapter. Phenoxide is resonance-stabilised; alkoxide is not. Substituents on the phenol ring shift the pKa predictably.
Electron-withdrawing groups (NO2, X, CN) at ortho or para: stabilise the phenoxide via resonance and -I, raising acidity. p-Nitrophenol (pKa 7.1) is roughly 1000 times more acidic than phenol.
Three -NO2 groups at 2, 4, 6 positions: give picric acid (2,4,6-trinitrophenol) with pKa 0.4, stronger than acetic acid. Picric acid preparation: phenol -> p-nitrophenol (dil. HNO3) -> 2,4-dinitrophenol (more conc. HNO3) -> picric acid (conc. HNO3 + H2SO4). The PDF works the full three-step preparation.
Electron-donating groups (CH3, OCH3) at ortho or para: destabilise the phenoxide, lowering acidity. p-Cresol (pKa 10.2) is slightly less acidic than phenol.
Meta-position EWGs: only contribute through inductive effect, not resonance. m-Nitrophenol (pKa 8.4) is more acidic than phenol but less than p-nitrophenol.
Ether Cleavage with HI, Bromination of Phenol, Friedel-Crafts on Phenol
Three electrophilic-substitution and cleavage reactions account for 70% of phenol/ether questions. The PDF derives each.
Ether cleavage HI: R-O-R' + HI gives R-I + R'-OH (smaller alkyl becomes the iodide via SN2). For anisole + HI, the products are phenol + methyl iodide (NEVER iodobenzene + methanol) because the aryl-oxygen bond resists cleavage from partial double-bond character. For methyl tert-butyl ether + HI, the products are methanol + tert-butyl iodide via SN1 at the tertiary carbon.
Bromination of phenol: phenol + Br2 in water gives 2,4,6-tribromophenol (white precipitate, no catalyst needed). In CS2 / CHCl3 at low temperature, only mono-substitution gives ortho- and para-bromophenol. The aqueous Br2 reaction is also a qualitative test for phenol.
Friedel-Crafts on phenol: works but yields are poor because -OH coordinates AlCl3. Anisole reacts cleanly. p-Acyl-anisole is the major product since -OCH3 is ortho/para-directing (+M effect dominates).
Saytzeff dehydration: conc. H2SO4 dehydration of 2-methylbutan-2-ol at 443 K gives 2-methylbut-2-ene as the major product (more substituted alkene, Saytzeff rule). The CBSE 3-mark mechanism question on dehydration asks for this regiochemistry.
Alcohol Oxidation: PCC vs KMnO4 vs Cu Dehydrogenation
Oxidation reagent choice is the most-tested 2-mark question in this chapter. The PDF tabulates which oxidant stops where.
Substrate
PCC (mild)
KMnO4 / K2Cr2O7 (vigorous)
Cu, 573 K (dehydrogenation)
1 degree R-OH
R-CHO (stops at aldehyde)
R-COOH (goes to acid)
R-CHO + H2
2 degree R-OH
R2C=O (ketone)
R2C=O (ketone)
R2C=O + H2
3 degree R-OH
No reaction
C-C cleavage (carboxylic acid mixture)
Alkene (no alpha-H, dehydrates)
PCC in CH2Cl2 is the only reagent that stops a primary alcohol at the aldehyde stage without going further to acid. This is the 1-mark MCQ pattern in NEET 2024.
Esterification Mechanism (Fischer Esterification)
NCERT works the Fischer esterification mechanism in section 7.6. The PDF reproduces the six-arrow mechanism: (i) protonation of the carbonyl O, (ii) nucleophilic addition of R-OH to give the tetrahedral intermediate, (iii) proton transfer from the alkoxy O to the -OH, (iv) loss of water, (v) deprotonation, (vi) regeneration of the catalyst H+. The equilibrium is driven by removing water (Dean-Stark trap in the lab).
For Ar-OH (phenol), Fischer esterification is sluggish because the phenolic lone pair is delocalised into the ring; the Schotten-Baumann route (acid chloride + NaOH or pyridine) is preferred.
Class 12 Chemistry Chapter-wise Marks Distribution (CBSE 2026-27)
Where does Chapter 7 sit in the bigger picture? The bar profile below shows approximate marks for every Class 12 Chemistry chapter under the 2026-27 blueprint, so you can decide your revision sequence by yield-per-hour.
Chapter
Topic
Approx. CBSE Marks
Ch 1
Solutions
5
Ch 2
Electrochemistry
5
Ch 3
Chemical Kinetics
5
Ch 4
The d- and f-Block Elements
4
Ch 5
Coordination Compounds
5
Ch 6
Haloalkanes and Haloarenes
4
Ch 7
Alcohols, Phenols and Ethers
7
Ch 8
Aldehydes, Ketones and Carboxylic Acids
8
Ch 9
Amines
5
Ch 10
Biomolecules
4
Chapters 7, 8, and 9 together carry roughly 20 marks, which is why the organic block deserves the lion's share of revision time after the inorganic chapters are wrapped up.
Related Resources for Class 12 Chemistry Chapter 7
All NCERT Solutions for Alcohols, Phenols and Ethers with Step-by-Step Working
Every NCERT textbook question for Class 12 Chemistry Chapter 7 Alcohols, Phenols and Ethers is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
Questions
Q 7.1
Write IUPAC names of the following compounds:
(i) CH3-CH(CH3)-CH(OH)-C(CH3)2-CH3
(ii) CH3-CH(OH)-CH2-CH(OH)-CH(C2H5)-CH2-CH3
(iii) CH3-CH(OH)-CH(OH)-CH3 (iv) HO-CH2-CH(OH)-CH2-OH
(v) 2-methyl-6-hydroxy substituted benzene
(vi) 4-methylphenol (vii) 3-methylphenol with OH at C-2
(viii) 2,6-dimethylphenol (ix) CH3-O-CH2-CH(CH3)-CH2-CH3
(x) C6H5-O-C2H5 (xi) C6H5-O-C7H15 (n-)
(xii) CH3-CH2-O-CH(CH3)-CH2-CH3.
Concept used.IUPAC nomenclature for alcohols
and ethers proceeds in three fixed steps. First, find the longest
continuous carbon chain that carries the -OH group; this is
the parent alkane and the -OH replaces a final ``-e'' with
``-ol''. Second, number the chain so that the carbon bearing
-OH gets the lowest possible locant (-OH has priority
over alkyl branches and halogens for low numbering). Third, list
the substituents alphabetically with their locants as prefixes.
For ethers we name the smaller R-O- group as an
alkoxy substituent on the longer carbon chain (the
parent). For phenols, the benzene ring carrying -OH is named
``phenol'' and -OH is at C-1 by default.
Priority of suffix groups
When more than one functional group is present, the principal
characteristic group is chosen by the IUPAC priority list. Between
-OH (suffix ``-ol'') and alkyl branches, -OH wins. So
locant 1 is assigned to give the lowest number to the carbon
bearing -OH.
(i) The skeleton is
CH3-CH(CH3)-CH(OH)-C(CH3)2-CH3. The longest chain
containing -OH has 5 carbons (pentane), and the
-OH sits on the middle (C-3) carbon. Both
numbering directions therefore give the same locant (3)
to the principal group. The tie is broken by ``lowest
locants for substituents at the first point of
difference''. Numbering from the gem-dimethyl end gives
two methyls on C-2 and one methyl on C-4, i.e. locant
set 2,2,4; numbering from the other end gives
2,4,4. Set 2,2,4 wins at the second locant
(2 < 4). Final name:
2,2,4-trimethylpentan-3-ol.
(ii) The skeleton
CH3-CH(OH)-CH2-CH(OH)-CH(C2H5)-CH2-CH3 has the
longest -OH-containing chain of 7 carbons: number
from the left to keep both OH groups low. Locants are
C-2 and C-4 (diol set 2,4 is lower than 4,6
from the right). An ethyl group sits at C-5. Final name:
5-ethylheptane-2,4-diol.
(iii) CH3-CH(OH)-CH(OH)-CH3. Four-carbon
chain (butane) with OH at C-2 and C-3. Name:
butane-2,3-diol.
(iv) HO-CH2-CH(OH)-CH2-OH. Three-carbon
chain (propane) with three OH groups at C-1, C-2, C-3.
Name: propane-1,2,3-triol (common name:
glycerol).
(v) A benzene ring with -OH at C-1 and a
-CH3 at the adjacent ortho carbon (C-2). Name:
2-methylphenol (o-cresol).
(vi) Benzene ring with -OH at C-1 and
-CH3 at C-4. Name: 4-methylphenol
(p-cresol).
(vii) Benzene ring with -OH at C-1 and a
-CH3 at C-3. Name: 3-methylphenol
(m-cresol).
(viii) Benzene ring with -OH at C-1 and
methyls at C-2 and C-6. Name: 2,6-dimethylphenol.
(ix) CH3-O-CH2-CH(CH3)-CH2-CH3. The longer
side of the ether oxygen is the butyl chain
(-CH2-CH(CH3)-CH2-CH3); the shorter side is
-OCH3 (methoxy). Numbering the parent butane from
the end nearer the alkoxy gives OCH3 at C-1, methyl
at C-2. Name: 1-methoxy-2-methylbutane.
(x) C6H5-O-C2H5 = ethoxybenzene. Treat the
phenyl side as the larger parent (benzene). The smaller
ethyl-O side is the alkoxy substituent. Name:
ethoxybenzene (common: phenetole).
(xi) C6H5-O-(CH2)6-CH3 (n-heptyl).
Phenoxy substituent on heptane: 1-phenoxyheptane. But
IUPAC also accepts naming benzene as the parent when the
substituent chain is acyclic. Standard NCERT answer:
1-phenoxyheptane.
(xii) CH3-CH2-O-CH(CH3)-CH2-CH3. The longer
side of the oxygen is -CH(CH3)-CH2-CH3 (a 3-carbon
chain with a methyl branch on C-1 of the parent end);
the shorter is -OC2H5 (ethoxy). The parent (after
choosing the longer carbon side) is butane via the
chain CH3-CH(O Et)-CH2-CH3 where the OC2H5 is
the alkoxy substituent on butane at C-2. Name:
2-ethoxybutane.
Structural observation. Every name above follows the
same template: parent chain length + suffix for the principal
group + locants for substituents. The trick is to identify the
parent chain correctly when more than one chain length is
possible, and to recognise that the priority of the principal
characteristic group (-OH) overrides the priority of any
mere substituent (alkyl, halogen).
Alternative approach: ``three-step decoder''. For any
compound name, (1) circle the suffix and its locant, (2) find
the parent chain or ring that bears it, (3) attach all
substituents at their locants. The reverse procedure works for
naming: tag the principal group, find the longest chain through
it, then label substituents.
For (i), the parent is pentane (5 C), with the
-OH on the middle carbon (C-3). Because OH sits at
the central carbon, its locant is 3 regardless of
numbering direction. The substituent locants decide
the tie: numbering from the gem-dimethyl end gives
2,2,4 for the three methyls, the other way gives
2,4,4. ``Lowest locants at the first point of
difference'' picks 2,2,4. Hence
2,2,4-trimethylpentan-3-ol.
For (ii), pick the longest chain that includes
both OH groups. The continuous chain runs through
all seven carbons of the main backbone, giving heptane.
Numbering from the methyl end nearer the first OH places
OH at C-2 and C-4. An ethyl branch sits at C-5. Final
name: 5-ethylheptane-2,4-diol. The
``first point of difference'' rule resolves the
diol-locant set 2,4 vs 4,6 in favour of the
former.
For polyols (iii), (iv) the parent name keeps the
terminal ``-e'' before ``-diol''/``-triol'' to avoid two
consecutive vowels colliding: ``butane-2,3-diol'', not
``butan-2,3-diol''. The same rule will apply to amines
(``-diamine'') in the next chapter.
For aromatic compounds (v) to (viii) the ring is the
parent, -OH is at C-1, and the locants are chosen so
the methyl substituents get the smallest numbers. The
common names o/m/p-cresol are still widely accepted
in industry but IUPAC nomenclature is required in
exams.
For ethers (ix) to (xii), apply the substitutive-ether
rule: name the smaller side as
``R-oxy'' (-OR) and treat it as a substituent on
the larger parent. ``Methoxy'' = -OCH3, ``ethoxy'' =
-OC2H5, ``phenoxy'' = -OC6H5. For
C6H5-O-C7H15 (xi) the longer ``side'' would be the
heptane chain (7 C > benzene's effective 6 C in
substitutive nomenclature), so heptane is the parent and
phenoxy is the substituent.
Concept linkage. The IUPAC priority list places
-OH (suffix ``-ol'') below -COOH, -CHO, C=O
and a few others, but above amines, ethers, halogens and alkyl
branches. So if both -OH and -COOH sit on the same
molecule, the acid takes the suffix and the alcohol becomes
``hydroxy'' as a prefix. You'll need this hierarchy whenever
biomolecules (Ch 14, e.g. carbohydrates with -OH and -CHO) are
named.
Exam relevance. JEE Main and CBSE board exams typically
allocate one MCQ or one 2-mark question to IUPAC nomenclature of
alcohol-ether mixtures; expect at least one such question
every year. Common trap: not picking the longest chain that
contains -OH (test case is exactly (i) above).
Why this matters. A clean IUPAC name lets a chemist
re-draw the structure unambiguously: it is the working language
of every later mechanism, spectroscopy or retrosynthesis problem.
Spectroscopists also use it as the key to look up reference
NMR/IR data.
Names as listed in the main solution.
Q 7.2
Write structures of the compounds whose IUPAC names are
as follows:
(i) 2-Methylbutan-2-ol (ii) 1-Phenylpropan-2-ol
(iii) 3,5-Dimethylhexane-1,3,5-triol
(iv) 2,3-Diethylphenol (v) 1-Ethoxypropane
(vi) 2-Ethoxy-3-methylpentane
(vii) Cyclohexylmethanol (viii) 3-Cyclohexylpentan-3-ol
(ix) Cyclopent-3-en-1-ol (x) 4-Chloro-3-ethylbutan-1-ol.
Concept used. To go from an IUPAC name to a structure,
reverse the naming algorithm: identify the parent chain length
from the root (but, pent, hex, etc.), place the principal group
at its locant, and attach each substituent on its locant carbon.
(i) 2-Methylbutan-2-ol. Parent: butane (4 C).
OH at C-2; methyl at C-2 as well. Structure:
CH3-C(OH)(CH3)-CH2-CH3.
(ii) 1-Phenylpropan-2-ol. Parent: propane.
OH at C-2; phenyl at C-1. Structure:
C6H5-CH2-CH(OH)-CH3.
(iii) 3,5-Dimethylhexane-1,3,5-triol. Parent:
hexane. OH at C-1, C-3, C-5; methyls at C-3, C-5.
Structure:
HO-CH2-CH2-C(CH3)(OH)-CH2-C(CH3)(OH)-CH3.
(iv) 2,3-Diethylphenol. Benzene with OH at C-1;
ethyl groups at C-2 and C-3:
o-(C2H5)-m-(C2H5)-C6H3-OH.
(v) 1-Ethoxypropane. Parent: propane. Ethoxy
-OC2H5 at C-1. Structure:
CH3-CH2-CH2-O-CH2-CH3.
(vi) 2-Ethoxy-3-methylpentane. Parent: pentane.
Ethoxy at C-2, methyl at C-3. Structure:
CH3-CH(OC2H5)-CH(CH3)-CH2-CH3.
(vii) Cyclohexylmethanol. A -CH2OH on
cyclohexane: C6H11-CH2OH.
(viii) 3-Cyclohexylpentan-3-ol. Parent: pentane.
OH and a cyclohexyl group both at C-3:
CH3-CH2-C(C6H11)(OH)-CH2-CH3.
(ix) Cyclopent-3-en-1-ol. Cyclopentene with the
double bond between C-3 and C-4 and an -OH at C-1.
Structure: a 5-membered ring with one C=C two carbons
away from the C bearing OH.
(x) 4-Chloro-3-ethylbutan-1-ol. A 4-carbon
parent labelled C-1 to C-4 starting from the -OH;
ethyl at C-3, Cl at C-4. Structure:
HO-CH2-CH2-CH(C2H5)-CH2Cl. (NCERT names this with
-CH2Cl terminal as ``4-chloro-3-ethylbutan-1-ol''
even though strict IUPAC would re-number.)
(i)–(x) structures as drawn above.
PS
Priya Sharma
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Picture-first. The easiest way is to draw the parent
skeleton with numbered carbons, then ``decorate'' it with
substituents and the principal group. The opposite of Q 7.1: we
go from name to structure, so we read the locant of the
``-ol'' (or ``-en-ol'') first, then add substituents.
Alternative approach: build it like LEGO. Start with
the parent block (a n-carbon chain or ring numbered 1 to n),
snap on the principal group at its locant, then click on each
substituent in turn. For polyols, place all OH groups before
methyls or ethyls so you do not lose count.
For each item, draw the parent: n carbons in a row,
labelled C-1, C-2, , C-n left to right (or as a
ring for cyclic parents). Use bond-line shorthand if
you wish: the ends and corners are carbons, hydrogens
are implicit.
Attach the suffix group (the ``-ol'' or ``-en-ol'') at
its locant carbon. Remember that ``en'' designates a
C=C double bond between two specified carbons.
Add substituents at their locant carbons. For (iv) the
phenol carbon is C-1 (carries OH), then go ortho to it
for C-2 and continue around the ring for C-3.
For (ix), the double bond ``3-en'' specifies a
C3=C4 bond, two carbons away from the OH at C-1.
That gives the symmetric cyclopent-3-en-1-ol.
For (x), 4-chloro-3-ethylbutan-1-ol has an apparent
conflict: butan-1-ol's parent has only 4 carbons but
``3-ethyl'' adds 2 more (making 6 total). The 4-carbon
parent is chosen because it carries the principal
group -OH at C-1; the ethyl branch sits on C-3
and the chloro on C-4.
Concept linkage. Reading a name backwards is the same
skill set as drawing organic products from a reaction equation.
Both require breaking the name into root, suffix and prefixes,
and rebuilding the connectivity on paper.
Exam relevance. Almost every CBSE Class 12 chemistry
paper has a 1-mark ``draw the structure of X'' question
somewhere; the trick is to identify the parent length from the
root (but, pent, hex) and the principal group from the suffix
(-ol, -al, -oic acid, -one).
Numerical sanity check. The molecular formula
of each compound should match. For (iii)
3,5-dimethylhexane-1,3,5-triol, count: C8H18O3
(6 C in hexane + 2 methyl branches = 8 C; 3 OH + the rest is
saturated → 18 H + 3 O). Cross-check with the drawn structure.
Why this matters. This decode skill is exactly what an
exam reverse-name question tests: parse the name into pieces,
then re-build the molecule piece by piece. The same skill lets
you convert a reaction product like ``2-bromo-2-methylbutane''
into a drawable structure quickly (Q 7.33).
(i)–(x) structures as drawn above.
Q 7.3
(i) Draw the structures of all isomeric alcohols of
molecular formula C5H12O and give their IUPAC names.
(ii) Classify the isomers of alcohols in question 7.3 (i) as
primary, secondary and tertiary alcohols.
Concept used. For C5H12O (a saturated, acyclic
formula C_nH_2n+2O), all isomers with an -OH group on
sp3 carbon are pentanol-type alcohols. An alcohol is
classified as primary (1∘) if its -OH-bearing
carbon is attached to one other C; secondary (2∘)
if to two other C; tertiary (3∘) if to three. The
carbon-skeleton isomers of pentane are three: n-pentane,
isopentane (2-methylbutane), neopentane (2,2-dimethylpropane). On
each skeleton, the OH can sit at any chemically distinct carbon.
n-pentane skeleton (CH3-CH2-CH2-CH2-CH3):
OH can go on C-1 (= pentan-1-ol, 1∘),
C-2 (= pentan-2-ol, 2∘), or
C-3 (= pentan-3-ol, 2∘).
2-methylbutane skeleton
(CH3-CH(CH3)-CH2-CH3): OH can go on
C-1 (= 2-methylbutan-1-ol, 1∘),
C-2 (= 2-methylbutan-2-ol, 3∘),
C-3 (= 3-methylbutan-2-ol, 2∘),
or on the terminal of the methyl branch =
3-methylbutan-1-ol, 1∘.
2,2-dimethylpropane skeleton: OH on a CH3
gives (CH3)3C-CH2OH = 2,2-dimethylpropan-1-ol
(neopentyl alcohol), 1∘. No other distinct
position on this skeleton.
Strategic angle. Walk through the three carbon
skeletons of C5 (pentane, 2-methylbutane,
2,2-dimethylpropane). On each, mark all chemically distinct
carbons and place -OH on each in turn. This ``skeleton
→ substitution-site'' algorithm is the cleanest way to
enumerate structural isomers.
Alternative approach: degree-of-unsaturation check.
C5H12O has DoU = (25 + 2 - 12)/2 = 0. So all eight
isomers are saturated, acyclic, and have one -OH (no rings, no
double bonds). This rules out, e.g. a five-membered ring with
an OH (which would have DoU = 1).
Pentane has 3 distinct carbons by symmetry (C-1 = C-5,
C-2 = C-4, C-3): so 3 OH positions → 3 alcohols.
Pentan-1-ol is 1∘; pentan-2-ol and pentan-3-ol are
both 2∘.
2-Methylbutane has 4 distinct carbons (the three on the
main chain plus the methyl branch): so 4 OH positions
→ 4 alcohols. 2-Methylbutan-1-ol and
3-methylbutan-1-ol are 1∘; 3-methylbutan-2-ol is
2∘; 2-methylbutan-2-ol is 3∘ (only 3∘
isomer in the set).
2,2-Dimethylpropane (neopentane) has 2 distinct carbons
(the central C and the four equivalent methyls): only
one OH position gives a valid alcohol (on a methyl); OH
on the central C would replace a methyl, which is not a
substitution but a different skeleton. So 1 alcohol:
2,2-dimethylpropan-1-ol (neopentyl alcohol), 1∘.
Concept linkage. The same enumeration trick is used in
Q 7.7 (C7H8O phenols/alcohols) and in the haloalkanes
chapter (C5H11Br, 8 isomers). The pattern ``one functional
group + all distinct skeletons + all distinct substitution
positions'' gives every constitutional isomer.
Exam relevance. Counting isomers is a perennial 1- or
2-mark question. The trap is to over-count by treating an
already-counted structure as new (e.g., the so-called
``4-methylbutan-1-ol'' is just 2-methylbutan-1-ol reversed). Use
the canonical IUPAC name as a unique key.
Numerical aside. The number of acyclic alcohol isomers
of C_nH_2n+2O grows quickly: n=11, n=21,
n=32, n=44, n=58, n=617.
Why this matters. Counting structural isomers by
``skeleton then substitution position'' is the cleanest method
and generalises to halides, amines and ethers. It is also a
prerequisite for spectroscopy: when an NMR shows ``six signals,
one CHOH at δ 3.7'', that already pins one of the eight
isomers down.
Eight isomers in total: 4 primary, 3 secondary,
1 tertiary.
Q 7.4
Explain why propanol has higher boiling point than that
of the hydrocarbon, butane.
Concept used. Boiling point depends on the strength of
intermolecular forces that must be broken to take a
liquid to the gas phase. For comparable molecular masses, the
ranking of these forces is
hydrogen bonding > dipole-dipole > London dispersion. Hydrogen bonding (H-bonding) is an unusually strong
dipole-dipole attraction between an -OH (or -NH) on one
molecule and a lone pair of an electronegative atom (O, N, F) on
a neighbouring molecule.
Compare molecular masses.
M(C3H7OH) = 3(12) + 7(1) + 16 + 1 = 60 g/mol;
M(C4H10) = 4(12) + 10(1) = 58 g/mol. The
two molecules have nearly the same mass and similar size.
Identify forces in butane (C4H10). Butane is
non-polar, so its only intermolecular force is weak
London dispersion (induced dipole-induced
dipole). Boiling point: -0.5.
Identify forces in propanol (C3H7OH). Propanol has
an -OH group, which provides a polar O-H bond
and a lone pair on oxygen. So propanol molecules form
hydrogen bonds between an O-H of one molecule
and the lone pair of O on another:
R-O-H ⋯ O(H)-R.
Each H-bond is worth roughly 20kJ/mol, much
stronger than London dispersion (1–10kJ/mol).
Boiling point of propan-1-ol: 97.
Conclusion. A vapourising propanol molecule must break
several hydrogen bonds, while a vapourising butane
molecule only needs to break dispersion contacts. So
propanol has a much higher boiling point.
[See diagram in the PDF version]
Propanol's molecules associate through hydrogen
bonding (an extra ∼20 kJ/mol attraction per pair) while
butane only has weak London forces, so much more energy is
needed to vapourise propanol than butane.
AV
Aditi Verma
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Anchor the comparison in numbers: both
molecules weigh about 60 g/mol, yet their boiling points differ
by nearly 100. Such a gap can only come from a
qualitatively different intermolecular force–-hydrogen bonding
is the only candidate.
Alternative approach: enthalpy of vaporisation comparison.
A clean way to make the case is to look up Δ Hvap:
Δ Hvap(butane) = 22.4kJ/mol,
Δ Hvap(propan-1-ol) = 47.5kJ/mol.
The gap of 25kJ/mol is the energetic cost of breaking
roughly one or two hydrogen bonds per molecule on
vaporisation–-a direct measurement of the H-bond energy.
Butane is a non-polar alkane. Its only attractions are
London dispersion, which scale with surface area and
polarisability. At 58 g/mol they give a b.p. of
-0.5 (about 273 K).
Propanol has an -OH group. The O-H bond is highly
polar (Δχ = 1.24 on the Pauling scale), so
δ+ on H and δ- on O. The H on one
molecule attracts the O lone pair on a neighbour: a
hydrogen bond. The geometry of the H-bond is almost
linear (∠O–H⋯O ≈ 175∘) and
the O⋯O distance is about 2.8.
Hydrogen-bond strength: about 20kJ/mol per
O-H O contact. Each propanol molecule can act
once as donor and twice as acceptor (oxygen has two lone
pairs), forming an associated, chain-like cluster in
the liquid. In propan-1-ol's solid phase, X-ray data
show extended H-bonded helices.
To vapourise, the molecule must escape this cluster.
That extra cost of breaking H-bonds raises the b.p. to
97 (370 K), almost 100
above butane.
Concept linkage: alcohol vs alkane vs ether. An ether
(e.g. methoxymethane, C2H6O) has the same skeleton size
as propanol but no O-H bond. Its boiling point is
-24, far below propanol's 97.
So the lesson generalises: ``OH bonded to electronegative atom''
is the differentiator, not just ``contains O''. Q 7.22 explores
this with ethanol vs dimethyl ether.
Exam relevance. Boiling-point comparison questions
(∼1–2 marks) appear in every CBSE paper. The expected
answer must (1) name the intermolecular force in each compound,
(2) rank their strengths, and (3) connect to b.p. Stating
``hydrogen bonding'' alone is half-marks; you must also explain
why butane cannot.
Numerical aside.Kb-style intuition: a 25 kJ/mol gap
in Δ Hvap predicts a b.p. gap by Trouton's
rule Tb ≈ Δ Hvap/85 J K-1 mol-1
⇒ Δ Tb ≈ (25 000/85) ≈ 290 K
in idealised cases. In practice the gap is smaller
(100) because Trouton's rule overestimates
strongly-associated liquids.
Why this matters. Hydrogen bonding is the single biggest
reason organic chemistry treats alcohols, amines and water-like
solvents very differently from alkanes. The same logic explains
why DNA strands hold each other (H-bonds between bases) and why
ice floats on water (the H-bond network of solid ice is less
dense than liquid water).
Propan-1-ol forms hydrogen bonds; butane does not.
Hence propanol has the higher boiling point (+97 vs
-0.5 ∘C).
Q 7.5
Alcohols are comparatively more soluble in water than
hydrocarbons of comparable molecular masses. Explain this fact.
Concept used. A solute dissolves in a solvent when the
solute-solvent attractions are comparable to (or stronger than)
both the solute-solute and the solvent-solvent attractions that
must be broken. For water (H2O), the solvent-solvent
attraction is hydrogen bonding. So a solute that can form
hydrogen bonds with water dissolves, while one that cannot is
forced out (the ``hydrophobic effect'').
Identify the H-bonding sites in an alcohol. Each
R-OH has one O-H (donor) and two lone pairs on O
(acceptors). So an alcohol can both donate and
accept hydrogen bonds with water.
Identify them in a hydrocarbon. A pure hydrocarbon
(e.g. propane, butane) has only C-H bonds. C-H is
almost non-polar; H bonded to C cannot serve as an
H-bond donor, and there is no lone pair to accept either.
Compare solvation energies. Dissolving propan-1-ol in
water replaces water-water H-bonds with new
R-O-H O(H2) and R-O H-OH
contacts, which are roughly the same strength. So the
process is thermodynamically near-neutral, i.e.
propan-1-ol is miscible with water. Butane, on the
other hand, can only offer weak dispersion forces to
the water network and so it is forced into a separate
layer (almost insoluble).
Trend with chain length. As the alkyl tail grows, the
hydrophobic part dominates over the hydrogen-bonding
-OH, and solubility falls: methanol and ethanol
are fully miscible, but hexan-1-ol is only sparingly
soluble.
[See diagram in the PDF version]
Alcohols form hydrogen bonds with water (R-O-H acts
as donor; lone pair on O acts as acceptor). Hydrocarbons cannot
form such bonds, so they are far less soluble than alcohols of
comparable molecular mass.
VR
Vivaan Reddy
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Structural observation. Two requirements for a small
molecule to dissolve in water are (a) polarity, and (b) the
ability to form hydrogen bonds. Alcohols meet both; hydrocarbons
meet neither.
Alternative approach: thermodynamic accounting.
Dissolution is governed by Δ Gsoln = Δ
Hsoln - TΔ Ssoln. For alcohols
Δ Hsoln ≈ 0 (water-water H-bonds replaced
by water-alcohol H-bonds of similar strength); Δ
Ssoln>0 (mixing entropy); so Δ Gsoln < 0
and the alcohol dissolves. For hydrocarbons Δ H ≈ 0
but Δ Ssoln<0 (the ``iceberg'' of ordered water
around the hydrophobe), so Δ Gsoln>0 and they
do not dissolve.
In water, every H2O is surrounded by roughly four
H-bonded neighbours (tetrahedral arrangement). To insert
a guest molecule, some of these water-water H-bonds must
be temporarily broken (cost: ∼20 kJ/mol each).
An alcohol pays this cost back: it forms new
water-alcohol H-bonds of similar strength
(∼20 kJ/mol). Net enthalpy of solution is small;
entropy of mixing is favourable; so it dissolves. For
ethanol, methanol, propan-1-ol, water-miscibility is
complete (1:1 in all proportions).
A hydrocarbon offers no replacement H-bonds. Water
molecules near the hydrocarbon are forced into a more
ordered ``cage'' (lower entropy: Δ S contribution
of -30 to -50 J/(mol K) for small alkanes). Net
free energy of solution is positive (Δ G ≈
+10 kJ/mol for butane), so the hydrocarbon is excluded:
the hydrophobic effect.
Longer-chain alcohols (hexan-1-ol, heptan-1-ol)
increasingly behave like hydrocarbons because the
chain length overwhelms the single -OH. Standard
solubility data:
MeOH, EtOH, PrOH: miscible;
BuOH: 8.0 g/100 g water;
PentOH: 2.2 g/100 g;
HexOH: 0.6 g/100 g.
Concept linkage: phenol vs alcohol solubility. Phenol
(pKa ∼ 10) is moderately soluble in water (8 g/100 g at
20); above 66 it becomes fully
miscible. The phenolic OH H-bonds with water, but the aromatic
ring is largely hydrophobic. Compare this with octan-1-ol, which
is almost insoluble in water despite having an OH. Solubility is
always a battle between the H-bonding head and the hydrophobic
tail.
Exam relevance. Solubility questions are usually
phrased as comparisons. The expected answer always cites
(1) intermolecular forces in the pure solute, (2) forces between
solute and water, and (3) the net thermodynamic balance.
Numerical hook. The Hildebrand solubility parameter
δ provides a quick way to predict miscibility:
δ(water) = 47.8 J1/2 cm-3/2,
δ(ethanol) = 26.5, δ(hexane) = 14.9.
Compounds with similar δ values are miscible; large gaps
mean phase separation. Hexane and water differ by 32 units–-
they are immiscible.
Why this matters. The ``like dissolves like'' rule of
thumb is really shorthand for matching intermolecular forces.
Water dissolves what it can H-bond with; oil dissolves what it
can only dispersion-bond with. This is the basis of soap action,
membrane biology, and even why colours in your laundry detergent
work–-each is a balance between hydrophilic and hydrophobic
parts of the same molecule.
The -OH of an alcohol forms hydrogen bonds with
water; a hydrocarbon cannot. Hence alcohols are far more
water-soluble than hydrocarbons of comparable mass.
Q 7.6
What is meant by hydroboration-oxidation reaction?
Illustrate it with an example.
Concept used. The hydroboration-oxidation
reaction is a two-step transformation that converts an alkene
into a primary alcohol with anti-Markovnikov
regiochemistry. Step 1 (hydroboration): diborane
(B2H6, equivalent to BH3) adds across the C=C
double bond in a single concerted, syn-addition; boron goes to
the less-substituted carbon, hydrogen to the more-substituted
carbon. The product is a trialkylborane, R3B.
Step 2 (oxidation): alkaline hydrogen peroxide
(H2O2 in aqueous NaOH) replaces the C-B bond by a
C-OH bond with retention of configuration. The overall result is
formal addition of H-OH to the alkene with H on the more
substituted carbon and OH on the less substituted carbon, the
opposite of acid-catalysed hydration (Markovnikov).
Step 1, hydroboration. The boron atom in BH3 has
only six valence electrons, so it is electrophilic. The
π-electrons of the alkene attack boron; the boron-H
bond then breaks, delivering H to the other carbon.
Because the B-H adds to one face of the alkene in a
single transition state, the addition is syn.
Three such additions consume one BH3 to give a
trialkylborane R3B.
3 RCH=CH2 + BH3 -> (RCH2CH2)3B.
Step 2, oxidation. Treat the trialkylborane with
alkaline H2O2:
(RCH2CH2)3B + 3 H2O2 OH- 3 RCH2CH2OH + B(OH)3.
The peroxide oxygen displaces the alkyl group from
boron, then water delivers the proton to give the
alcohol.
Example. Start from propene
(CH3-CH=CH2). Hydroboration places B on C-1
(less substituted) and H on C-2 (more substituted):
CH3-CH=CH2 (i) B2H6 (CH3-CH2-CH2)3B.
Oxidation gives propan-1-ol:
(CH3-CH2-CH2)3B (ii) H2O2, OH- CH3-CH2-CH2-OH + B(OH)3.
Notice that acid-catalysed hydration of the same
propene would have given propan-2-ol (Markovnikov),
so hydroboration-oxidation gives the
opposite regiochemistry.
[See diagram in the PDF version]
Hydroboration-oxidation = (i) addition of B2H6
across an alkene; (ii) oxidation by alkaline H2O2 to
give an anti-Markovnikov primary alcohol. Example: propene
→ propan-1-ol.
RK
Rohit Kapoor
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Frame the reaction as a two-step
``protection-deprotection'' of the alkene's two carbons: boron
labels the less-substituted carbon, then H2O2 swaps the
label for OH. The boron is a temporary marker that tells the
oxygen where to land.
Alternative approach: Markovnikov vs anti-Markovnikov
choice. For an alkene RCH=CH2, you have two main ways to
add ``water'':
Acid hydration (H2SO4/H2O): Markovnikov,
OH on the more-substituted carbon (gives a 2∘
alcohol from a terminal alkene).
Hydroboration-oxidation (B2H6/H2O2, OH-):
anti-Markovnikov, OH on the less-substituted carbon (gives
a 1∘ alcohol from a terminal alkene).
Choose based on which alcohol regiochemistry your target
demands.
In step 1, BH3 approaches the alkene with the
empty p-orbital pointing at the π cloud. Steric
bulk forces boron onto the less-substituted end of the
double bond–-this regiochemistry is set by the
transition state, not by any electronic preference (the
π-bond is symmetric; the steric clash with the
substituent decides).
Three equivalents of alkene react with one BH3
(because BH3 has three B-H bonds), producing a
trialkylborane. The C-B bond is essentially non-polar
(electronegativities ∼2.0 vs 2.5), so the
intermediate is moderately stable and isolable.
The addition is syn: H and B end up on the same face
of the alkene–-useful for stereochemistry.
In step 2, the hydroperoxide anion HOO- (from
H2O2 in NaOH) adds to boron; an alkyl group
migrates from B to O with retention of configuration at
carbon; water then hydrolyses the B-O-R bond to
give R-OH and boric acid (B(OH)3). The migration
is the key step and is one of the rare cases where C-B
rearranges to C-O.
For propene, the net change is propene →
propan-1-ol (yield 90–95%). Compare with H3O+
hydration, which gives propan-2-ol (Markovnikov,
2∘ alcohol). For 2-methylpropene
((CH3)2C=CH2), hydroboration gives the
1∘ alcohol (CH3)2CH-CH2-OH
(2-methylpropan-1-ol), while acid hydration would give
the 3∘(CH3)3C-OH.
Concept linkage: protection-deprotection strategy.
Hydroboration-oxidation is one of the few additions to alkenes
with anti-Markovnikov regiochemistry. Other anti-Markovnikov
methods include peroxide-mediated HBr addition (radical
mechanism). Knowing both Markovnikov and anti-Markovnikov
methods doubles the synthetic toolkit available for any alcohol
target.
Exam relevance. ``Convert alkene X to alcohol Y''
questions test exactly this regiochemistry choice. The dead
giveaway is: target is 1∘ alcohol from terminal
alkene ⇒ hydroboration-oxidation;
target is 2∘ or 3∘ alcohol from terminal alkene
⇒ acid hydration.
Numerical aside. Stereochemistry yield: in
hydroboration of a chiral alkene, both faces are accessible but
only one ``syn'' product forms per face attack, leading to a
racemate. The reaction is stereospecific (syn addition) but not
enantioselective with achiral BH3.
Why this matters. This reaction is the standard way to
make a primary alcohol from a terminal alkene: it is the
synthetic complement of acid hydration. H. C. Brown won the
1979 Nobel Prize for developing this and other organoborane
reactions. In modern synthesis it remains the cleanest way to
install OH at a primary position.
Hydroboration-oxidation = anti-Markovnikov ``hydration''
of an alkene; propene → propan-1-ol via syn-addition of
B-H followed by retention-of-configuration oxidation.
Q 7.7
Give the structures and IUPAC names of monohydric
phenols of molecular formula C7H8O.
Concept used. A monohydric phenol has one
-OH group attached directly to a benzene ring. The formula
C7H8O contains seven carbons; subtracting the six in the
ring leaves one extra carbon, which must be a methyl group on
the ring. The methyl group can occupy the ortho (C-2),
meta (C-3) or para (C-4) position relative to
the -OH. We also note that ``benzyl alcohol''
(C6H5-CH2-OH) has the same molecular formula but is not
a phenol because its -OH is on the side-chain carbon, not
on the ring.
2-methylphenol (ortho-cresol): a
benzene ring with -OH at C-1 and -CH3 at C-2.
3-methylphenol (meta-cresol): benzene
ring with -OH at C-1 and -CH3 at C-3.
4-methylphenol (para-cresol): benzene
ring with -OH at C-1 and -CH3 at C-4.
[See diagram in the PDF version]
Three monohydric phenols of formula C7H8O:
2-methylphenol, 3-methylphenol and 4-methylphenol.
TB
Tara Banerjee
M.Sc Chemistry, IIT Kanpur
Verified Expert
Structural observation. For a monohydric phenol we
need one -OH on the benzene ring. With seven carbons in
total and six in the ring, exactly one carbon remains as a
ring substituent: it has to be -CH3. The only freedom is
its ring position.
Alternative approach: degree of unsaturation. For
C7H8O, DoU = (27 + 2 - 8)/2 = 4. Four
degrees fit one benzene ring (DoU = 4) and nothing else. So
every isomer is a benzene derivative; the remaining one carbon
must sit as a CH3 (or be incorporated into the ring as part
of a 7-membered ring, but cyclooheptatrienol is not stable and
is not a monohydric phenol).
Fix -OH at C-1. Three distinct positions remain
for the methyl: C-2 (ortho), C-3 (meta), C-4 (para).
Positions C-5 and C-6 are equivalent by symmetry to
C-3 and C-2 respectively, so they are not separate
isomers.
Each of the three isomers (ortho, meta, para) is a
named compound, called o-, m- and p-cresol respectively.
They are real-world chemicals found in coal tar and
creosote.
Confirm that no other phenol-type isomer exists: a
seven-carbon phenol must place the seventh carbon as
a one-carbon side chain (since two-carbon ones would
give C8H10O). So three is the complete count.
For completeness, the only non-phenol isomer of
C7H8O that is also an aromatic alcohol is benzyl
alcohol (C6H5-CH2-OH), which has the OH on the
side chain, not the ring. It is excluded from this
question's count.
Concept linkage: alcohol vs phenol distinction. The OH
in an alcohol is on a sp3 carbon; the OH in a phenol is on a
sp2 (aromatic) carbon. This difference shapes everything:
acidity (pKa ∼ 16 vs ∼ 10), reactions with NaOH
(no for alcohol, yes for phenol), reactions with NaHCO3
(no for both, mostly), and reactions with HX (alcohol →
alkyl halide; phenol does not).
Exam relevance. Counting questions (``how many isomers
of formula X are alcohols/phenols/ethers'') are standard 1- or
2-mark items in CBSE and JEE. The trap is always benzyl
alcohol or other side-chain isomers that the student may
mistakenly include in a phenol count.
Spectroscopic distinguisher.1H NMR of phenols shows
the OH proton at δ 4–8 (very variable, depending on
solvent and concentration) and a strongly downfield-shifted
broad signal. The aromatic protons of cresols cluster around
δ 6.8–7.0. Comparing chemical shifts of the three
cresols lets us spot ortho/meta/para directly.
Why this matters. Cresols are industrial disinfectants
(Lysol is a mixture of these isomers). They also illustrate
ortho/meta/para classification, the workhorse of aromatic
substitution. m-Cresol is used in resin manufacture
and is a precursor to vitamin E synthesis.
Three: 2-, 3- and 4-methylphenol (the o-, m-, p-cresols).
Q 7.8
While separating a mixture of ortho and
para nitrophenols by steam distillation, name the
isomer which will be steam volatile. Give reason.
Concept used. A compound is steam volatile if
it has appreciable vapour pressure at 100
(the temperature of boiling water) and does not associate
strongly with water. Strong intermolecular hydrogen bonding
between solute molecules lowers vapour pressure and prevents
steam volatility. Intramolecular hydrogen bonding
within a single molecule, in contrast, locks up the -OH
internally and stops intermolecular association, leaving the
molecule free to vapourise. Hence the isomer with
intramolecular H-bonding is the steam-volatile one.
Look at the geometry. In ortho-nitrophenol, the
-OH at C-1 sits right next to the -NO2 at C-2.
The O-H hydrogen can swing across to form a hydrogen
bond with one of the -NO2 oxygens within the same
molecule (a 6-membered chelate ring).
In para-nitrophenol, the -OH (C-1) and the
-NO2 (C-4) are diametrically opposite on the ring.
Their distance is too large for intramolecular
H-bonding. Instead, each -OH forms intermolecular
H-bonds with neighbouring molecules' -NO2 groups,
giving an extended, associated network.
Consequence for vapour pressure. Para-nitrophenol
forms a strongly H-bonded solid (m.p. 114,
b.p. 279). Ortho-nitrophenol's
intramolecular bond replaces some intermolecular ones, so
it has weaker overall lattice forces (m.p.
45, b.p. 216).
Result. Ortho-nitrophenol passes over with the
steam; para-nitrophenol stays behind. Steam
distillation therefore separates them.
[See diagram in the PDF version]
Ortho-nitrophenol is steam-volatile because
its -OH forms an intramolecular hydrogen bond with the
adjacent -NO2, lowering intermolecular association.
Para-nitrophenol is held in an intermolecular
H-bonded network and stays behind.
AP
Aanya Pillai
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Steam distillation works for a
compound whose vapour pressure plus that of water reaches
1 atm at ≤ 100. Anything tied up in a
strong intermolecular hydrogen-bonded network has too low a
vapour pressure to do this. The decisive factor is whether
H-bonding is internal (favours volatility) or external
(hinders volatility).
Alternative approach: melting-point comparison. A
quick lab proxy for ``how associated is this solid'' is the
melting point: o-nitrophenol melts at 45,
m-nitrophenol at 97, p-nitrophenol at
114. The lowest-melting isomer has the
weakest lattice forces, exactly what you need for steam
volatility.
In o-nitrophenol the OH and NO2 are on adjacent
carbons; the O-H bond points toward an -NO2 oxygen
across a six-membered ring. The intramolecular hydrogen
bond ``saturates'' the OH so it cannot form many
intermolecular contacts. The ring closure for the
H-bond involves 6 atoms (O-H⋯O-N-C-C), the most
stable ring size for chelation.
In p-nitrophenol no such intramolecular contact is
geometrically possible (OH at C-1 and NO2 at C-4
are at opposite ends, separated by ∼5.8).
Each -OH forms two or more intermolecular bonds:
the solid is held together like a 3-D polymer of
H-bonded units. Crystal-structure data confirm head-to-
tail ribbons in p-nitrophenol crystals.
Vapour pressures at 100: o-isomer high
enough to co-evaporate with water (about 1 mmHg);
p-isomer essentially zero on this scale (<0.01 mmHg).
Steam distillation exploits Dalton's law of partial
pressures: total pressure = pwater +
psolute. Boiling occurs when total = 1 atm.
In the laboratory, the o-isomer condenses in the
receiving flask (bright yellow crystals); the p-isomer
is recovered from the distillation residue (also
yellow but more deeply coloured).
Concept linkage: chelation rings. The same
intramolecular H-bond stabilisation appears in salicylaldehyde
(2-hydroxybenzaldehyde, where OH and CHO chelate), in
2-nitroaniline (NH2 and NO2 chelate), and in
β-diketones (enol form chelates). The 6-membered
chelate ring is a recurring stabilisation motif throughout
organic chemistry.
Exam relevance. The o/p-nitrophenol separation is a
standard 2–3 mark question. Always (1) draw both isomers,
(2) circle the intramolecular H-bond in the ortho, (3) explain
why this lowers intermolecular association, and (4) connect
to vapour pressure / b.p. Just saying ``ortho has H-bond, para
doesn't'' is half-marks; the reasoning must connect.
Acidity paradox. The same intramolecular H-bonding
also makes o-nitrophenol less acidic than the p-isomer
in water: the OH proton is partly tied up in the chelate
and harder to release. pKa: o-7.23, p-7.15. A subtle
0.08 unit gap but real.
Why this matters. Steam distillation is a non-trivial
separation technique that exploits volatility differences
caused by H-bonding. It is used industrially to purify
essential oils (citral, eugenol) and to extract
heat-sensitive natural products that would decompose at their
true boiling point.
The ortho isomer is steam volatile due to
its intramolecular H-bond (chelation, 6-membered ring);
the para isomer is held back by intermolecular
H-bonding.
Q 7.9
Give the equations of reactions for the preparation
of phenol from cumene.
Concept used. The cumene process (Hock
process) is the industrial route to phenol. Cumene
is isopropylbenzene, C6H5-CH(CH3)2. The benzylic
C-H of cumene is easily oxidised by atmospheric oxygen to
a hydroperoxide, which rearranges in acid to give
phenol and acetone. The reaction is industrially attractive
because both products (phenol and acetone) are valuable.
Step 1: prepare cumene by Friedel-Crafts alkylation of
benzene with propene over an acid catalyst:
C6H6 + CH3-CH=CH2 H+ C6H5-CH(CH3)2.
Step 2: aerial oxidation. Pass air through cumene at
∼120 in the presence of a small
amount of acid; the tertiary benzylic C-H abstracts an
oxygen molecule to give cumene hydroperoxide:
C6H5-CH(CH3)2 + O2 -> C6H5-C(CH3)2-OOH.
Step 3: acid-catalysed rearrangement (Hock
rearrangement). Treat the hydroperoxide with dilute
acid; the O-O bond breaks with migration of the
phenyl group:
C6H5-C(CH3)2-OOH H3O+ C6H5-OH + CH3-CO-CH3.
The two products are phenol and acetone.
Cumene is regenerated industrially by alkylating
benzene with the acetone-derived propene (after
dehydration), making the overall process near-circular.
Strategic angle. Track the carbon skeleton: benzene
(6 C) plus propene (3 C) gives cumene (9 C); cumene splits
back into phenol (6 C) and acetone (3 C). The propene carbons
end up in acetone, the benzene carbons in phenol. This atom
economy makes the process attractive: every C from the feed
ends up in a useful product.
Alternative approach: thermodynamic driving force.
The Hock rearrangement is exergonic by about
-90kJ/mol–-driven by formation of two strong C=O
bonds (in phenol's enol tautomer briefly, and in acetone) at
the cost of one O-O bond (a weak ∼150kJ/mol) and
one C-C bond. Without this large negative Δ G, the
rearrangement would not be spontaneous.
The C-H at the benzylic carbon of cumene is weak
(∼370kJ/mol) because the resulting
tertiary benzylic radical is stabilised by both
hyperconjugation and resonance with the ring. Compare
with a typical alkane C-H (∼410kJ/mol).
Air abstracts that hydrogen; the resulting
radical traps O2 to form a peroxy radical,
which picks up another H from a fresh cumene
molecule (radical chain propagation). Net product:
cumene hydroperoxide.
In acid, the OH of the peroxide is protonated; water
leaves; the phenyl group migrates from C to O (a
1,2-aryl shift). The oxocarbenium intermediate adds
water and breaks down to phenol plus a protonated
acetone, which tautomerises to acetone. This C-to-O
migration is the heart of the Hock rearrangement.
Yield of phenol per mole of cumene is essentially
quantitative (98–99%); both products are isolated by
fractional distillation. Acetone fractions out at
56 (atmospheric) and phenol at
182.
Concept linkage. The Hock rearrangement is a special
case of the broader Bayer-Villiger-like family of
``migration to electron-deficient oxygen'' reactions. The
migrating group tends to be the one that best supports a
partial positive charge during migration: aryl > tertiary
alkyl > secondary > primary > methyl. In cumene
hydroperoxide, phenyl migrates faster than methyl, giving
phenol selectively.
Exam relevance. ``Preparation of phenol from cumene''
is a guaranteed 2–3 mark question. Always (1) write all
three reagent/condition equations, (2) name the
co-product (acetone), and (3) mention that the process is
industrially dominant. Bonus: name the Hock rearrangement.
Yield numerical. If 1 mole of cumene (120 g/mol) gives
1 mole of phenol (94 g/mol) at 95% yield, the mass yield is
94 × 0.95 / 120 = 0.74 g phenol per g cumene. Industrial
plants routinely achieve this benchmark.
Why this matters. The Hock rearrangement is one of
the few large-scale industrial migrations of an aryl group
from carbon to oxygen, exploited because of the value of both
products. About 95% of the world's phenol (12 million
tonnes/year) and a major share of acetone (6 million tonnes/year)
are produced this way.
Write chemical reaction for the preparation of
phenol from chlorobenzene.
Concept used.Chlorobenzene
(C6H5Cl) is very unreactive in normal nucleophilic
substitution because the C-Cl bond has partial double-bond
character from π-donation by the chlorine lone pair into
the ring. To force the substitution, harsh conditions are
needed. The industrial Dow process uses
6–8 aqueous NaOH at
623K (∼350) and high pressure
(200–300 atm). The mechanism is the elimination-addition
``benzyne'' pathway.
Treat chlorobenzene with fused NaOH (or
8 aqueous NaOH at 623K
and 300atm) to give sodium phenoxide:
C6H5Cl + 2 NaOH 623K, 300 atm C6H5ONa + NaCl + H2O.
Acidify the resulting phenoxide salt with dilute
HCl (or H2SO4) to liberate phenol:
C6H5ONa + HCl -> C6H5OH + NaCl.
Strategic angle. The two-step nature of this synthesis
is typical of aromatic hydroxylations: first install O-Na
on the ring under harsh conditions, then neutralise with
acid to free phenol. The same form-salt/acidify motif appears in
the benzenesulphonate route (Q 7.12) and in the cumene process
indirectly.
Alternative approach: comparing the three industrial
routes to phenol.
Cumene (Q 7.9): mild conditions, two valuable products,
dominant route today (95% of global phenol).
Benzenesulphonate (Q 7.12): oldest route (1899),
sulphonation then fusion with NaOH.
Knowing all three lets you pick the right answer for any
specific exam question.
Under industrial conditions (623K, 300 atm),
the strong base OH- deprotonates a ring
hydrogen ortho to Cl, then Cl- leaves to give a
benzyne intermediate (a transient
sp2-sp ring with an in-plane π-bond).
OH- then adds to one of the two
triple-bonded carbons, giving phenoxide after proton
transfer. This elimination-addition mechanism explains
why isotopic labelling at the ortho carbon shows
scrambling.
The sodium phenoxide is water-soluble (the
C6H5O- ion is moderately stabilised by
ring resonance and by Na+ counter-ion) and is
extracted into the aqueous layer; chlorobenzene
(unreacted) and benzene byproducts go to the organic
layer.
Treatment of the phenoxide with HCl protonates
the oxygen to release neutral phenol, which is then
separated by distillation. The aqueous NaCl byproduct
is discarded.
Yield of phenol: about 80–85% on industrial scale
(historically Dow Chemical's flagship process before
the cumene route overtook it). The harsh conditions
and equipment cost made it unattractive once the
Hock chemistry was perfected.
Concept linkage: nucleophilic aromatic substitution.
The Dow process is the prototype of an
elimination-addition (SNAr via benzyne)
mechanism on an aryl halide. Direct SNAr (addition-
elimination) needs strong -M groups ortho/para to the halide
(see ChemDraw of p-NO2-C6H4-Cl + NaOH, which
goes by addition-elimination at lower temperature). Without
such activators, only benzyne works.
Exam relevance. The Dow process is a standard
1–2 mark question. Always (1) write the equation with
623K, 300 atm conditions, (2) note that you need a
strong base, and (3) include the acidification step. Optional
bonus: mention benzyne mechanism.
Why this matters. The Dow process illustrates how
``unreactive'' aryl halides become reactive under forcing
basic conditions via benzyne. It also shows the typical
two-step ``form-the-salt, then-acidify'' protocol of phenol
synthesis. The benzyne intermediate, demonstrated by Wittig
and Roberts in the 1950s, was a watershed in mechanistic
organic chemistry.
Write the mechanism of hydration of ethene to yield
ethanol.
Concept used.Acid-catalysed hydration of an
alkene is an electrophilic addition. The proton from
H3O+ (generated by H2SO4 in water) attacks the
π-bond first, giving a carbocation. Water then attacks the
carbocation as a nucleophile, and finally a base (water itself)
removes the extra proton from the oxocarbenium to give the
neutral alcohol. The reaction is reversible: low water-content
favours dehydration, high water-content favours hydration.
Step 1: protonation of the alkene. A water molecule
carrying a proton (H3O+ from H2SO4 in
water) attacks the π-electrons of ethene. The
proton adds to one carbon, leaving a primary
carbocation on the other:
CH2=CH2 + H3O+ <=> CH3-CH2+ + H2O.
This is the slow, rate-determining step.
Step 2: nucleophilic attack of water on the
carbocation. A second water molecule uses one of its
oxygen lone pairs to attack the empty p-orbital of
the cation:
CH3-CH2+ + H2O <=> CH3-CH2-OH2+.
This gives a protonated alcohol (an oxocarbenium ion).
Step 3: deprotonation. A third water molecule removes
the extra proton from the oxocarbenium, regenerating
H3O+ and giving neutral ethanol:
CH3-CH2-OH2+ + H2O <=> CH3-CH2-OH + H3O+.
The catalyst H3O+ is regenerated at the end, as
expected. Overall:
CH2=CH2 + H2O H2SO4 CH3-CH2-OH.
!%
[See diagram in the PDF version]
Violet curly arrows show electron-pair flow: π pair grabs H+ from H3O+; an O lone pair of water then attacks C+; finally a water base lifts H+ off the oxonium ion to deliver ethanol.
Three-step mechanism: protonation → carbocation
→ nucleophilic attack of water → deprotonation →
ethanol.
YB
Yash Bhat
M.Sc Chemistry, IIT Kanpur
Verified Expert
Structural observation. The intermediate is an
ethyl cation, CH3-CH2+, which is primary. The reaction
is slower than the corresponding hydration of propene (which
goes through the more stable secondary cation
CH3-CH+-CH3). For this reason, ethene needs higher
temperature and pressure than propene.
Alternative approach: Markovnikov framing. Although
ethene's two carbons are identical (by symmetry, no regiochemistry
question), the general acid-hydration mechanism is the textbook
Markovnikov example. For propene, the cation lands on C-2
(more substituted) and OH ends up there too–-this is the
mnemonic ``rich gets richer''. For ethene, the symmetry makes
the issue trivial.
In a typical industrial setup, ethene is mixed with
98 H2SO4 at 300
and 70 atm. The first step is protonation of the
alkene to form CH3-CH2+. The acid donor in
concentrated H2SO4 is actually H3SO4+
(protonated sulphuric acid) or the equivalent
H3O+ in dilute conditions.
The proton donor under these conditions is actually
H3O+ or H2SO4 itself; both work the
same way mechanistically. The slow step is this
protonation (activation energy 120kJ/mol for
ethene; only 90kJ/mol for propene–-hence the
rate gap).
Once the carbocation is formed, water (the bulk
solvent) traps it rapidly: the lone pair on O attacks
the empty p-orbital of C+. This is barrierless
within the diffusion limit for primary carbocations.
The protonated alcohol is then deprotonated by another
water molecule to give ethanol and regenerate
H3O+. Le Chatelier's principle: an excess of
water pushes the equilibrium toward ethanol; an excess
of H2SO4 at higher T reverses it to ethene
(Q 7.19).
Concept linkage: hydration vs hydroboration-oxidation.
Acid hydration: Markovnikov; cation mechanism; works best for
non-terminal or branched alkenes (stable 2∘ or 3∘
cation). Hydroboration-oxidation (Q 7.6): anti-Markovnikov;
concerted; works best for terminal alkenes when a 1∘
alcohol is desired. Both deliver the same atoms but to
opposite carbons.
Exam relevance. The mechanism of ethene hydration is
a 3–4 mark CBSE question. The complete answer must show
(1) protonation, (2) cation formation, (3) water attack, and
(4) deprotonation, with curly arrows for each step. Skipping
the third water (the proton remover) is a common deduction.
Numerical context. Industrial conversion per pass:
about 5%. The unreacted ethene is recycled. Worldwide
production of ``synthetic'' ethanol (from ethene rather than
sugar fermentation): about 5 million tonnes/year. Total
ethanol (sugar + synthetic) production is over 100 million
tonnes/year.
Why this matters. This is one of the workhorse
industrial routes to ethanol; understanding the mechanism is
the foundation for the broader topic of electrophilic addition
to alkenes (HX, X2, hypohalous acids), all of which
follow the same protonation-cation-trap-deprotonation logic.
You are given benzene, conc. H2SO4 and
NaOH. Write the equations for the preparation of phenol
using these reagents.
Concept used. This is the benzenesulphonate
fusion route to phenol. Sulphonate the benzene ring with
conc. H2SO4 to install -SO3H; neutralise to the
sodium sulphonate; then fuse with solid NaOH at high
temperature so that the -SO3^- group is displaced by
-O^-, giving sodium phenoxide. Acidify to free phenol.
Sulphonation. Heat benzene with concentrated
H2SO4. The electrophile SO3 (or the
protonated form) substitutes a ring H:
C6H6 + H2SO4 Δ C6H5-SO3H + H2O.
The product is benzenesulphonic acid.
Neutralisation. Treat the sulphonic acid with
NaOH to make the salt:
C6H5-SO3H + NaOH -> C6H5-SO3Na + H2O.
Alkali fusion. Heat solid sodium benzenesulphonate
with solid NaOH at ∼573K–623K:
C6H5-SO3Na + 2 NaOH Δ C6H5-ONa + Na2SO3 + H2O.
The strong base displaces the sulphonate (a nucleophilic
aromatic substitution under forcing conditions).
Acidification. Dissolve the sodium phenoxide in water
and acidify with dilute HCl (the conjugate acid
of H2O, or even CO2/water in industry):
C6H5-ONa + HCl -> C6H5-OH + NaCl.
Strategic angle. The plan is electrophilic
substitution (to install -SO3H) followed by harsh
nucleophilic substitution (to swap -SO3Na for -ONa).
Both SO3^- and Cl^- can be ``forced off'' an aromatic
ring at high temperature with strong base, but SO3^- is
the better leaving group of the two–-hence why this route
uses milder conditions than the Dow process (Q 7.10).
Alternative approach: comparing leaving groups on
benzene. The classic ``hard-to-displace'' aromatic leaving
groups are arranged in increasing order of how easily they
leave when fused with NaOH: -H < -NH3+ < -Cl
< -Br < -SO3- < -N2+. So sulphonate
is a useful leaving group at moderately high T, while
-H never leaves directly.
Conc. H2SO4 at 40–60
sulphonates benzene; the active electrophile is
SO3 (or its protonated form HSO3+).
This is reversible (heating with dilute acid would
reverse it), which is actually exploited in the
``ipso protection'' strategy for selective EAS on
complex aromatics.
The free acid is converted to its sodium salt by
neutralisation with aqueous NaOH. The sodium
sulphonate is highly water-soluble and is easily
isolated by evaporation.
Alkali fusion is done in solid state at high temperature
(573–623K) because aqueous OH-
alone is not strong enough for SNAr without an
activator on the ring. The molten NaOH generates
an aggressive ``naked'' O2- equivalent, which
attacks the aromatic carbon, expelling sulphite. The
byproduct Na2SO3 goes into the aqueous wash.
Final acidification with dilute HCl liberates
phenol; it is extracted into an organic solvent
(typically ether or chloroform) and purified by
distillation (b.p. 182).
Concept linkage: three routes for phenol.
Sulphonation route (this question): historical,
still viable for small-scale lab synthesis with the
reagents at hand (benzene, H2SO4, NaOH).
Dow route (Q 7.10): chlorobenzene + NaOH; harsh
but uses only one harsh step (no sulphonation).
Cumene route (Q 7.9): industrial dominant; mild
conditions, two products.
Exam relevance. The exact question prompt names the
three reagents: benzene, conc. H2SO4, and NaOH.
You must use all three. Forgetting the final
acidification step (which actually requires a fourth reagent,
HCl) costs marks; some marking schemes accept CO2/water
or even no extra reagent (the phenoxide is acidic enough to
be displaced by carbonic acid).
Why this matters. The route demonstrates that even
``unreactive'' aromatic positions can be functionalised if you
choose the right leaving group and apply enough heat. The
BASF (Germany) process used this route from 1899 until the
1950s–-it was the workhorse of European phenol production
before the cumene era.
Four steps: sulphonation, salt formation, alkali
fusion, acidification; net C6H6 -> C6H5OH.
Q 7.13
Show how will you synthesise:
(i) 1-phenylethanol from a suitable alkene.
(ii) cyclohexylmethanol using an alkyl halide by an SN2
reaction.
(iii) pentan-1-ol using a suitable alkyl halide.
Concept used. Three different alcohol syntheses:
acid-catalysed Markovnikov hydration of an alkene (i),
nucleophilic substitution of an alkyl halide by -OH^- via
SN2 (ii), and an indirect route through a Grignard reagent
or through dilution of an aldehyde via reduction (iii). Pick
the simplest disconnection for each.
(i) 1-Phenylethanol from an alkene.
1-Phenylethanol is C6H5-CH(OH)-CH3. The
Markovnikov hydration of styrene
(C6H5-CH=CH2) places the OH on the more
substituted carbon (the benzylic one), giving exactly
this alcohol:
C6H5-CH=CH2 + H2O H2SO4 C6H5-CH(OH)-CH3.
(ii) Cyclohexylmethanol by SN2.
Cyclohexylmethanol is C6H11-CH2-OH. Start from
cyclohexylmethyl bromide (the primary halide
C6H11-CH2-Br) and treat it with aqueous
NaOH:
C6H11-CH2-Br + OH- SN2 C6H11-CH2-OH + Br-.
The primary halide undergoes a clean back-side
SN2 displacement.
(iii) Pentan-1-ol from an alkyl halide.
Pentan-1-ol is CH3-CH2-CH2-CH2-CH2-OH. Use
1-bromopentane and aqueous NaOH:
CH3-(CH2)3-CH2-Br + OH- SN2 CH3-(CH2)3-CH2-OH + Br-.
Primary 1∘ halides give the cleanest SN2
reactions (least competition from E2/SN1).
(i) Hydrate styrene with dilute H2SO4;
(ii) treat C6H11-CH2-Br with aq. NaOH;
(iii) treat C5H11-Br with aq. NaOH.
SG
Siddharth Gupta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Three syntheses, each illustrating a
different disconnection: (i) C-O bond from a π-bond, (ii)
C-O bond from a C-X bond, (iii) same idea as (ii) on a longer
chain. Pick the reagent that gives the target with no
isomerisation or rearrangement.
Alternative approach: retrosynthetic disconnection.
For any alcohol R-OH, disconnect at the C-O bond:
Disconnection A: R+ + OH-. Source of
R+: cation from alkene + H+ (Markovnikov path).
Disconnection B: R- + electrophilic O
(rare in practice).
For (i), styrene's hydration is Markovnikov because
the benzylic cation C6H5-CH+(CH3) is
resonance-stabilised by the ring (3 resonance forms
delocalise charge over o/p carbons). So the OH lands
on the carbon bonded to the ring, exactly as required.
The 1-phenylethanol product has pKa ∼ 15,
slightly more acidic than a simple alcohol due to the
benzylic anion's resonance.
For (ii), the back-side attack of OH- on the
primary carbon of C6H11-CH2-Br is a textbook
SN2; the cyclohexyl ring is bulky enough to slow
down any SN1 alternative (the secondary cyclohexyl
cation is much less stable than the primary
cyclohexylmethyl), leaving SN2 dominant. The
SN2 rate is about 103 times faster than for
comparable secondary halides.
For (iii), 1-bromopentane is a primary halide; aq.
NaOH at moderate temperature gives the alcohol
cleanly. Tertiary halides would not work here
(they would lose HBr to give an alkene instead via E2).
Yield of pentan-1-ol: typically 80–90%.
In all three cases, the byproducts (NaBr,
excess water, or starting alkene) are easily removed
by aqueous workup followed by simple distillation.
Concept linkage: when to use Grignard. For
(iii) pentan-1-ol, an alternative Grignard route is
CH3(CH2)3-MgBr + HCHO. This is sometimes preferred
when 1-bromopentane is more expensive than 1-bromobutane.
For (i) 1-phenylethanol, the Grignard alternative is
CH3MgBr + C6H5-CHO. Both routes give the same product;
choose by reagent cost and chain-length compatibility.
Exam relevance. ``Synthesise X from Y'' is a standard
multi-mark question. Always (1) state the reagent + conditions,
(2) write the equation with the correct SN2 / Markovnikov
notation, (3) name the mechanism explicitly.
Why this matters. The three reactions cover the two
big strategies for installing a C-O bond: oxymetalation/
hydration of an alkene, and nucleophilic substitution on a
halide. Together with Grignard addition (Q 7.20), these are
the entire alcohol-synthesis toolkit at NCERT level.
(i) Markovnikov hydration of styrene;
(ii) SN2 hydrolysis of C6H11-CH2-Br;
(iii) SN2 hydrolysis of CH3(CH2)4Br.
Q 7.14
Give two reactions that show the acidic nature of
phenol. Compare acidity of phenol with that of ethanol.
Concept used. Phenol behaves as a weak acid because
its O-H proton can be removed by a base, leaving a
phenoxide anion in which the negative charge is
delocalised over the ring carbons (resonance). The corresponding
conjugate base of ethanol, the ethoxide ion, has the
negative charge localised on oxygen (with destabilising
inductive donation from the ethyl group). So phenoxide is more
stable than ethoxide, which means phenol (pKa ≈ 10.0)
is much more acidic than ethanol (pKa ≈ 15.9).
Reaction 1, with aqueous NaOH. Phenol dissolves
in dilute NaOH giving a soluble sodium
phenoxide:
C6H5-OH + NaOH -> C6H5-ONa + H2O.
Ethanol does not react with cold dilute NaOH,
because ethanol's pKa (≈ 15.9) is comparable
to water's (≈ 15.7); the equilibrium does not
favour the ethoxide.
Reaction 2, with metallic sodium. Phenol releases
H2 on treatment with sodium metal:
2 C6H5-OH + 2 Na -> 2 C6H5-ONa + H2 .
Ethanol also gives this reaction, but more slowly:
2 CH3-CH2-OH + 2 Na -> 2 CH3-CH2-ONa + H2 .
Phenol fizzes vigorously; ethanol reacts steadily.
Comparison: in phenoxide, four resonance structures
place the negative charge on oxygen and on three ring
carbons (ortho, ortho, para). This delocalisation
lowers the energy of the phenoxide by tens of kJ/mol
relative to a localised charge.
In ethoxide CH3CH2O-, no such delocalisation
is possible; in fact the C2H5-group's inductive
+I effect destabilises the charge by pushing
electron density onto an already-negative oxygen.
So Ka(phenol)/Ka(ethanol) ≈
10(15.9 - 10.0) = 105.9 ≈ 8 × 105:
phenol is about a million times more acidic than
ethanol.
[See diagram in the PDF version]
Phenol reacts with NaOH and with Na
metal, releasing H2. Phenol (pKa ≈ 10) is about
105 times more acidic than ethanol (pKa ≈ 16)
because the phenoxide ion is resonance-stabilised whereas
ethoxide is not.
DC
Diya Chatterjee
M.Sc Physical Chemistry, IIT Madras
Verified Expert
Picture-first. Imagine the conjugate base in each case
and ask: where does the negative charge live? The answer to
this single question explains all the acidity behaviour.
Alternative approach: acidity ranking via inductive
and resonance effects. For any X-O-H system, the conjugate
base XO- is stabilised (and the acid X-OH made stronger)
by groups that withdraw electrons by -I or -M, and
destabilised by groups that donate by +I or +M. So:
Ethanol (C2H5-OH): ethyl group donates by +I,
destabilising ethoxide. Weak acid (pKa ∼ 16).
Water (H-OH): no substituent. Reference
(pKa = 15.7).
Phenol (C6H5-OH): aryl ring withdraws by -M,
stabilising phenoxide via resonance. Moderate acid
(pKa ∼ 10).
Acetic acid (CH3-COOH): C=O withdraws by
-I and -M, gives full resonance stabilisation.
Stronger acid (pKa ∼ 4.8).
In C2H5O- the charge sits on a single oxygen
atom with no neighbours that can share it. The
σ-only ethyl group cannot delocalise charge;
it actually destabilises the anion through its +I
effect (pushing more electron density onto the
already-negative O).
In C6H5O- the lone pair on oxygen overlaps
with the ring π-system; resonance moves the
negative charge onto carbons C-2, C-4, C-6 of the
ring (ortho, ortho, para positions). Four equivalent
resonance structures contribute: one with charge on O,
and three with charge on the ring carbons.
Spreading a charge over several atoms lowers its
free energy. So phenoxide is more stable than ethoxide;
equivalently, phenol holds onto its proton less
tightly than ethanol does. The free-energy difference
is about 34kJ/mol (from Δ pKa · RT
ln 10).
Quantitatively, pKa(phenol) = 10.0,
pKa(ethanol) = 15.9. So
Δ pKa = 5.9; phenol is roughly 105.9
≈ 8 × 105 times more acidic. Reactions:
phenol + NaOH goes to completion (Keq
= 1015.7-10.0 = 105.7); ethanol + NaOH
is essentially unreactive (Keq = 10-0.2).
Concept linkage: phenol vs alcohol vs ether. The
three oxygen-containing classes have very different
oxygen-pKa:
Ethers cannot ionise (no O-H bond) and are essentially
non-acidic.
Exam relevance. ``Why is phenol more acidic than
ethanol?'' is a classic 3-mark CBSE question. Full marks
require (1) draw both conjugate bases, (2) name the resonance
structures and inductive effects, (3) cite numerical pKa
values, and (4) explicitly compare with NaOH reactivity.
Numerical sanity check. The Ka of phenol is
10-10.0 = 1.0× 10-10 mol/L. In a 0.1 M phenol
solution, [H+] = √Ka C0 = √10-10·
0.1 = 10-5.5 M, giving pH ≈ 5.5. Compare with
ethanol: same concentration gives pH ≈ 8.4 (essentially
neutral).
Why this matters. The same logic explains why
p-nitrophenol (pKa ≈ 7.2) is more acidic
than phenol: the -NO2 group provides an additional
resonance sink for the negative charge. And why
p-cresol (pKa ≈ 10.3) is slightly less acidic:
the methyl group donates by +I/+H, destabilising the
phenoxide a little.
Phenol is acidic enough to react with NaOH;
ethanol is not. Resonance stabilisation of phenoxide (4 forms)
is the reason; phenol is ∼ 106 times more acidic.
Q 7.15
Explain why is ortho-nitrophenol more
acidic than ortho-methoxyphenol?
Concept used. The acidity of a substituted phenol
depends on how the substituent stabilises (or destabilises)
the resulting phenoxide. Two electronic effects matter:
inductive (-I) (electron withdrawal through
σ-bonds, stabilises the anion) and mesomeric
(-M resonance withdrawal or +M resonance donation through
π-bonds). The -NO2 group is strongly -I and -M
(both withdrawing), while -OCH3 is weakly -I but
strongly +M (donates π-density through the oxygen lone
pair).
Look at the phenoxide of o-nitrophenol. The
adjacent -NO2 pulls electron density toward
itself through both σ and π pathways,
dispersing the negative charge of the phenoxide ion
over the -NO2 oxygens as well. The conjugate
base is significantly stabilised. So
o-nitrophenol gives up its proton easily:
pKa ≈ 7.2.
Look at the phenoxide of o-methoxyphenol. The
-OCH3 group's oxygen has lone pairs which can
donate π-density into the ring (+M), adding
to the negative charge on the phenoxide oxygen. This
is destabilising. The -I effect of -OCH3 is
weak. Net effect: destabilisation. So
o-methoxyphenol is less acidic than
phenol itself: pKa ≈ 9.8.
Compare. Lower pKa = stronger acid. Since
pKa(o-NO2) = 7.2 <
pKa(o-OCH3) = 9.8,
o-nitrophenol is the stronger acid by a
factor of 102.6 ≈ 400.
Reason in one line: -NO2 withdraws electrons and
stabilises the negative phenoxide; -OCH3 donates
electrons and destabilises it.
o-Nitrophenol is more acidic because the
-NO2 group is an electron-withdrawing group (-I, -M)
that stabilises the phenoxide anion; whereas -OCH3 is
electron-donating (+M) and destabilises the anion.
MJ
Meera Joshi
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Structural observation. The key is to draw the
phenoxide ion in each case and ask whether the substituent
helps to spread the negative charge or works against it.
Alternative approach: substituent-effect calculator.
Use the Hammett σ-parameter table to estimate pKa
shifts:
For phenols, pKa = 10.0 - 2.2 p. So
pKa(p-NO2-phenol) ≈ 10.0 - 2.2(0.78)
≈ 8.3 (lit: 7.2; close enough). For p-OMe-phenol:
10.0 - 2.2(-0.27) ≈ 10.6 (lit: 10.2). The ortho data are
trickier because of steric and chelation effects, but the
direction is correctly predicted.
In o-nitrophenoxide, an extra resonance form
places the negative charge on an oxygen of the NO2
group. That extra delocalisation (5 resonance forms
total, vs 4 for plain phenoxide) makes the conjugate
base much more stable. The -I effect of the
NO2 group adds further σ-withdrawal.
In o-methoxyphenoxide, no such resonance form
is available. The OMe oxygen instead pumps lone-pair
density into the ring (the +M effect), increasing
electron density at the phenoxide oxygen, which is
already negative. Two negative charges on neighbouring
atoms repel; the conjugate base is destabilised.
Quantitatively, pKa values are 7.2 for
o-nitrophenol and 9.8 for
o-methoxyphenol; both compared to phenol's
10.0. So o-nitrophenol is 102.6 ≈
400 times more acidic than o-methoxyphenol.
Bigger picture: substituent effects on phenol acidity
sit on a sliding scale of -M/+M and -I/+I. -NO2
is the strongest -M group in NCERT, -OMe is a
moderate +M donor. The trend is general: EWGs lower
pKa, EDGs raise it.
Concept linkage: o vs m vs p. For the same substituent,
the order of acidity-modifying effect depends on position:
p position: full -M/+M resonance through the ring
(4 resonance structures involve the substituent).
o position: full -M/+M plus an inductive boost
from the proximity. Often the most acidifying (or
basifying) but sometimes complicated by chelation
(Q 7.8).
m position: only -I/+I matters; resonance does not
reach (no resonance form puts charge on the meta carbon).
For -NO2: p-nitrophenol pKa 7.2; m-nitrophenol
8.4; o-nitrophenol 7.2 (the o and p are similar, m is less
acidic because no resonance).
Exam relevance. Comparison-of-acidity MCQs are a
JEE staple. The decoder: bigger -M/-I at o/p→ more
acidic. NCERT tends to ask 4-option MCQs ranking
substituted phenols.
Why this matters. Predicting which phenol is more
acidic is a classic exam question that uses exactly this
substituent-effect logic; the same reasoning applies to
carboxylic acids and aromatic amines. The Hammett equation
quantifies the trend and is the basis of much of
physical-organic chemistry.
-NO2 stabilises the phenoxide anion (-I,
-M); -OCH3 destabilises it (+M). Hence
o-nitrophenol is the stronger acid (pKa 7.2 vs 9.8).
Q 7.16
Explain how does the -OH group attached to a
carbon of benzene ring activate it towards electrophilic
substitution?
Concept used. An electrophilic aromatic
substitution (EAS) proceeds by an electrophile E+
attacking the ring's π-cloud to form a positively charged
arenium ion (sigma complex), which then loses a
proton. The rate of EAS depends on how electron-rich the ring
is and how stable the arenium intermediate is. The -OH
group strongly activates the ring because its oxygen lone pair
donates π-density into the ring (+M, mesomeric donation),
making the ring more nucleophilic and the arenium intermediate
extra-stable.
Look at the ground-state ring. Lone pair of the OH
oxygen overlaps with the π-orbital of the ring,
pumping electron density to the ortho and para
carbons. The resonance structures place a δ-
on C-2, C-4 and C-6.
Look at the transition state. When an electrophile
E+ attacks at the ortho or para position, the
resulting arenium ion has a resonance form in which
the positive charge sits on the carbon bearing
OH; the lone pair on O then stabilises that positive
charge by forming an oxocarbenium-like resonance
structure (HO-C+ <=> HO+=C).
Because the OH lone pair shares the burden of
positive charge, the arenium ion is much more stable
than that of unsubstituted benzene. Lower activation
energy means a faster reaction.
Regiochemistry. The same resonance argument shows that
only ortho and para attack benefit from this
stabilisation: meta attack puts the + charge on
carbons not bonded to OH, so the oxygen lone pair
cannot help. Hence -OH is an ortho/para
director and a strong activator.
[See diagram in the PDF version]
The -OH oxygen's lone pair donates π-electron
density into the ring (+M), raising the HOMO of the ring and
stabilising the arenium intermediate at o/p positions. Hence
phenol is far more reactive than benzene toward EAS, especially
at o/p.
AR
Ananya Rao
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Activation = lowering the activation
energy of EAS. The OH group lowers it both in the starting
material (raises HOMO) and especially in the arenium ion
(directly donates to the cationic centre). The bigger
stabilisation in the transition state translates to a faster
reaction by the Hammond postulate.
Alternative approach: resonance structure count. For
unsubstituted benzene, the arenium ion from electrophilic
attack has 3 resonance forms. For phenol, attack at o/p gives
4 resonance forms including an oxocarbenium-like form with O
donating its lone pair. Attack at m gives only 3 forms (no
extra stabilisation). The 4-vs-3 ratio explains both the
activation (faster reaction) and the directing effect
(o/p preferred over m).
In phenol's resonance structures the OH oxygen has a
formal + charge and the ring carbon next to it has
a - charge: the ring is electron-rich at o/p. There
are three such ``charge-separated'' resonance forms
contributing to the ground-state stabilisation.
In the arenium ion formed by ortho or para attack,
the OH oxygen can donate its lone pair into the
positively charged ring system, forming an
``oxocarbenium'' resonance form that has all atoms
with full octets. This is the key stabilising form.
Activation energy of EAS at o/p is lowered by
∼30 kJ/mol compared to benzene.
Meta attack does not benefit from this donation because
the positive charge lands on carbons not adjacent to
the OH-bearing carbon. So m-EAS on phenol is only
marginally faster than on benzene, while o/p-EAS
is 105–106 times faster.
Result: phenol nitrates at room temperature with
dilute HNO3 to give o/p-nitrophenols (Q 7.17
iii); it brominates with Br2 in water to give
2,4,6-tribromophenol straight away (all three available
o/p positions get attacked). Compare benzene, which
requires conc. HNO3/H2SO4 at 60∘C
for mononitration.
Concept linkage: +M donors in EAS. The same
activation logic applies to -OR, -NH2, -NR2
substituents. All are strong +M donors and all are powerful
ortho/para directors. Rate enhancement vs benzene:
Exam relevance. ``Explain why phenol activates the
ring'' is a 3-mark CBSE question. Show
(1) ground-state resonance with charge-separated forms,
(2) arenium-ion resonance with oxocarbenium form,
(3) explicit mention of o/p (not m) and why,
(4) experimental comparison with benzene.
Why this matters. The same activation logic applies to
many synthetic problems. Knowing that phenol is far more
reactive than benzene also explains why protecting OH as OMe
(anisole) is sometimes needed (Q 7.31): plain phenol can
over-react (tri-bromo product instead of mono).
Lone pair on OH donates π-density into the ring;
arenium ion at o/p is extra-stabilised by an oxocarbenium
resonance form; phenol is therefore 105–106 times more
reactive than benzene in EAS.
Q 7.17
Give equations of the following reactions:
(i) Oxidation of propan-1-ol with alkaline KMnO4 solution.
(ii) Bromine in CS2 with phenol.
(iii) Dilute HNO3 with phenol.
(iv) Treating phenol with chloroform in the presence of aqueous
NaOH.
Concept used. Four different reactions of -OH
compounds: (i) alkaline KMnO4 is a strong oxidant that
takes a primary alcohol all the way to a carboxylic acid; (ii)
Br2 in CS2 at low temperature is a mild
brominating reagent for phenol, giving mainly para
monobromination; (iii) dilute HNO3 nitrates phenol at
room temperature, giving a mixture of o- and
p-nitrophenol; (iv) chloroform + aqueous NaOH
on phenol is the Reimer-Tiemann reaction, which
installs a -CHO group ortho to the OH.
(i) Alkaline KMnO4 on propan-1-ol gives
propanoic acid (passing through propanal as an
intermediate):
CH3-CH2-CH2-OH KMnO4, OH- CH3-CH2-COOH.
(ii) Phenol + Br2 in CS2 at low
temperature (273K) gives mainly
4-bromophenol (p-bromophenol). The
non-polar solvent (CS2) and low temperature
suppress polybromination:
C6H5-OH + Br2 CS2, 273Kp-HO-C6H4-Br + HBr.
(iii) Dilute HNO3 with phenol at room
temperature gives a mixture of o- and p-nitrophenols:
C6H5-OH + HNO3(dil.) -> o-NO2-C6H4-OH + p-NO2-C6H4-OH + H2O.
(iv) Reimer-Tiemann. Phenol with CHCl3
and aqueous NaOH at 340K, followed by
acidic workup, gives salicylaldehyde
(2-hydroxybenzaldehyde). The reactive
electrophile is dichlorocarbene :CCl2,
generated from CHCl3 + OH-:
aligned
CHCl3 + OH- &→ :CCl2 + Cl- + H2O,
C6H5-O- + :CCl2 &→ o-HOC6H4-CHO (after hydrolysis).
aligned
Strategic angle. Group the four reactions by type:
oxidation (i), electrophilic aromatic substitution (ii)+(iii),
and carbene insertion (iv).
(i) The C-H on the -OH-bearing carbon is
successively oxidised: -CH2OH → -CHO → -COOH.
With alkaline KMnO4, the reaction does not stop
at the aldehyde because the aldehyde is itself easily
oxidised. Acidic workup gives the free carboxylic acid.
(ii) Br2/CS2 at 273K is the textbook
condition for monobromination of phenol; para is the
major isomer due to less steric clash with the OH.
(iii) Dilute HNO3 has just enough NO2+
to nitrate phenol once; both o- and p- isomers form
and are separated by steam distillation (Q 7.8).
(iv) The Reimer-Tiemann reaction proceeds by attack
of dichlorocarbene at the ortho carbon of the phenoxide.
After alkaline hydrolysis of the -CHCl2 group,
you get the -CHO substituent.
Why this matters. These four reactions are the
``greatest hits'' of phenol chemistry: oxidation, ring
substitution and -CHO installation are all standard JEE
questions.
Equations as written in the main solution.
Q 7.18
Explain the following with an example.
(i) Kolbe's reaction.
(ii) Reimer-Tiemann reaction.
(iii) Williamson ether synthesis.
(iv) Unsymmetrical ether.
Concept used. Four named transformations or terms:
(i) the carboxylation of phenoxide by CO2 to give
salicylic acid; (ii) the formylation of phenol via
dichlorocarbene; (iii) the alkoxide + alkyl halide synthesis
of an ether; (iv) the definition of an ether with two
different R groups on oxygen.
(i) Kolbe's reaction. Treat sodium phenoxide
with CO2 at 400K and 4–7 atm; acidify.
The phenoxide attacks CO2 at the ortho carbon,
giving sodium salicylate, which on acidification
gives salicylic acid
(2-hydroxybenzoic acid):
!$C6H5-ONa + CO2 400 K, 4--7 atm 2-NaOOC-C6H4-OH H3O+ 2-HOOC-C6H4-OH$.
Salicylic acid is the precursor of aspirin.
(ii) Reimer-Tiemann reaction. Phenol +
CHCl3 + aqueous NaOH at 340K,
followed by acidic workup, gives
salicylaldehyde (2-hydroxybenzaldehyde). The
electrophile is dichlorocarbene
(:CCl2), made in situ from CHCl3 + OH-.
The carbene attacks the ortho carbon of the phenoxide;
the resulting -CHCl2 group is hydrolysed to
-CHO by the alkaline medium:
!$C6H5-OH CHCl3, NaOH, 340 K H3O+ o-HOC6H4-CHO$.
(iii) Williamson ether synthesis. React an
alkoxide (R-O-Na+) with a primary alkyl halide
(R′-X with X = Cl, Br, I). The alkoxide acts as a
nucleophile in an SN2 attack on the alkyl halide:
R-O- Na+ + R′-X → R-O-R′ + NaX.
Example: CH3-ONa + CH3-CH2-Br -> CH3-O-CH2-CH3 + NaBr
gives methoxyethane (methyl ethyl ether). The alkyl
halide must be primary or methyl; tertiary halides
give alkene by E2 instead.
(iv) Unsymmetrical (mixed) ether. An ether
R-O-R′ in which
R ≠ R′. Example:
ethyl methyl ether, CH3-O-C2H5. Versus
a symmetrical ether like CH3-O-CH3 (dimethyl
ether).
0.95!%
[See diagram in the PDF version]
(i) Kolbe: PhO-Na+ + CO2 -> salicylate;
(ii) Reimer-Tiemann: PhOH + CHCl3/NaOH -> salicylaldehyde;
(iii) Williamson: R-ONa + R′-X → R-O-R′;
(iv) Unsymmetrical ether: R-O-R′ with R ≠ R′.
RP
Riya Patel
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Three of these (i, ii, iii) are
named reactions on the JEE syllabus, each with a known
mechanism. The fourth (iv) is just a definition.
Kolbe's reaction works because the phenoxide ion is
electron-rich at ortho/para; it attacks the
electrophilic carbon of CO2 to form a new
C-C bond. The ortho carboxylate is thermodynamically
favoured under the reaction conditions.
Reimer-Tiemann uses the same activated phenoxide ring
as the nucleophile. The carbene :CCl2 inserts
at the ortho position, and aqueous NaOH
hydrolyses the dichloromethyl group to an aldehyde.
Williamson's SN2 step proceeds with inversion at
carbon. The alkoxide must be unhindered;
(CH3)3CO- does not attack primary alkyl
halides well because of steric crowding.
Unsymmetrical ethers (R-O-R′) are more useful
synthetic intermediates than symmetrical ones because
their two halves can be derived from two different
building blocks.
Why this matters. Together, Kolbe and Reimer-Tiemann
explain how Nature (and pharma) extracted aspirin and methyl
salicylate from phenol in the 19th century; Williamson is the
universal ether disconnection in modern synthesis.
Definitions and examples as written in the main
solution.
Q 7.19
Write the mechanism of acid dehydration of ethanol to
yield ethene.
Concept used.Acid-catalysed dehydration is
the reverse of acid-catalysed hydration (Q 7.11). It is an
E1 elimination passing through the same ethyl cation
intermediate. Concentrated H2SO4 at 443K
(∼170) gives the elimination product
(ethene); milder conditions (413K) give the
substitution product (diethyl ether), and even milder
(373K) give the ester ethyl hydrogen sulphate.
Step 1: protonation. Conc. H2SO4 protonates the
OH of ethanol to make a good leaving group
(H2O):
CH3-CH2-OH + H2SO4 <=> CH3-CH2-OH2+ + HSO4-.
Step 2: ionisation. Water leaves, forming the ethyl
cation:
CH3-CH2-OH2+ <=> CH3-CH2+ + H2O.
This is the slow, rate-determining step.
Step 3: deprotonation (E1). A base
(HSO4- or another water) abstracts a proton
from the carbon next to the cation, and the
two electrons of the C-H bond drop into the empty
p-orbital to form the new π-bond:
CH3-CH2+ + HSO4- <=> CH2=CH2 + H2SO4. H2SO4 is regenerated, confirming its catalytic
role.
Overall:
CH3-CH2-OH conc. H2SO4, 443 K CH2=CH2 + H2O.
!%
[See diagram in the PDF version]
E1 mechanism: (1) O lone pair grabs H+; (2) water leaves slowly to give the primary ethyl cation; (3) HSO4- (acting as a weak base) takes a β-proton while its C-H bonding pair (violet) drops into the empty p-orbital to form the new π bond of ethene.
Three-step E1 mechanism: protonation of OH →
loss of H2O to form CH3-CH2+→ loss of
proton from the adjacent carbon to give CH2=CH2.
NK
Neha Kumar
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. The reaction is Le Chatelier in
action: remove water (conc. H2SO4 is a desiccant) and
the equilibrium shifts to the alkene. Add water and the
alkene re-hydrates.
The first step is reversible protonation of the
alcohol. Sulphuric acid donates a proton to one of
the lone pairs on oxygen, converting -OH into the
much better leaving group -OH2+.
The second step is unimolecular ionisation: the
oxocarbenium loses water to give the ethyl cation.
This is rate-determining and accounts for the
``unimolecular'' label E1.
The third step is deprotonation of a β-carbon
by any base in solution (the HSO4-
counter-ion, water itself, or even another ethanol
molecule). The freed electron pair forms the π
bond.
Higher temperatures favour the elimination because
Δ S > 0 for E1 (two molecules of
product from one of reactant): elimination is
entropically favoured.
Why this matters. This is a textbook example of an
E1 mechanism: a carbocation intermediate, β-H
abstraction, no stereospecificity at β. Use it as the
template for all acid-catalysed alcohol dehydrations.
Mechanism: protonation → loss of water (slow,
gives CH3CH2+) → loss of β-H to give ethene.
Q 7.20
How are the following conversions carried out?
(i) Propene → Propan-2-ol.
(ii) Benzyl chloride → Benzyl alcohol.
(iii) Ethyl magnesium chloride → Propan-1-ol.
(iv) Methyl magnesium bromide → 2-Methylpropan-2-ol.
Concept used. Standard reagent-conversion problems.
(i) Markovnikov hydration of an alkene; (ii) hydrolysis of a
benzyl halide by aqueous NaOH (SN2); (iii) the
Grignard reagent adding to an aldehyde or a primary
epoxide to give a 1∘ alcohol; (iv) the same Grignard
reagent adding to a ketone to give a tertiary alcohol.
(i) Propene → Propan-2-ol. Treat propene
with dilute H2SO4 (or use
oxymercuration-demercuration for cleaner conditions);
Markovnikov adds OH to C-2:
CH3-CH=CH2 + H2O H2SO4 CH3-CH(OH)-CH3.
(ii) Benzyl chloride → Benzyl alcohol.
Reflux C6H5-CH2-Cl with aqueous NaOH or
aqueous Na2CO3:
C6H5-CH2-Cl + OH- SN2 C6H5-CH2-OH + Cl-.
The benzyl cation is stabilised by the ring; both
SN1 and SN2 pathways are accessible, but the
primary halide makes SN2 dominant.
(iii) C2H5MgCl → Propan-1-ol. Add
the Grignard to formaldehyde (HCHO); the ethyl
carbanion attacks the carbonyl carbon, then aqueous
workup gives the primary alcohol:
aligned
C2H5MgCl + HCHO &-> C2H5-CH2-OMgCl,
C2H5-CH2-OMgCl + H3O+ &-> C2H5-CH2-OH + Mg(OH)Cl.
aligned
Result: propan-1-ol CH3-CH2-CH2-OH.
(iv) CH3MgBr → 2-Methylpropan-2-ol.
2-Methylpropan-2-ol is (CH3)3C-OH. Add the
Grignard to acetone (propan-2-one,
CH3-CO-CH3); the methyl carbanion adds to the
ketone carbon, giving the tertiary alcohol after
workup:
aligned
CH3MgBr + CH3-CO-CH3 &-> (CH3)3C-OMgBr,
(CH3)3C-OMgBr + H3O+ &-> (CH3)3C-OH + Mg(OH)Br.
aligned
(i) Dil. H2SO4 + H2O;
(ii) aq. NaOH;
(iii) HCHO then H3O+;
(iv) CH3COCH3 then H3O+.
AB
Aditi Bhat
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Each conversion is one of four
classic ``install -OH'' strategies: hydration of an
alkene; hydrolysis of a halide; Grignard addition to an
aldehyde or to a ketone. Pick the right one for each starting
material.
(i) Markovnikov hydration. The 2∘ carbocation
CH3-CH+-CH3 is more stable than the primary
CH3-CH2-CH2+, so water adds to give
propan-2-ol (the 2∘ product).
(ii) Hydrolysis of benzyl chloride is rapid because
the benzyl carbon is electrophilic and primary
(SN2). Even mild base like Na2CO3 works.
(iii) Match the Grignard chain length with the
aldehyde so that the sum equals the chain length of
the target alcohol. C2H5MgCl (2 carbons) +
HCHO (1 carbon) = propan-1-ol (3 carbons).
(iv) For a 3∘ alcohol you need a ketone. Methyl
magnesium bromide (1 C) + acetone (3 C, two methyls
and a carbonyl) gives the tertiary
(CH3)3C-OH after workup.
Why this matters. These four conversions cover the
three big families of starting materials (alkenes, halides,
carbonyls) for making any saturated alcohol on a JEE/NEET
question paper.
(i) dil H2SO4/H2O;
(ii) aq NaOH;
(iii) HCHO, then H3O+;
(iv) CH3COCH3, then H3O+.
Q 7.21
Name the reagents used in the following reactions:
(i) Oxidation of a primary alcohol to carboxylic acid.
(ii) Oxidation of a primary alcohol to aldehyde.
(iii) Bromination of phenol to 2,4,6-tribromophenol.
(iv) Benzyl alcohol to benzoic acid.
(v) Dehydration of propan-2-ol to propene.
(vi) Butan-2-one to butan-2-ol.
Concept used. Each conversion tests recall of a
standard reagent. Strong oxidants take a 1∘ alcohol all
the way to a carboxylic acid; mild, selective oxidants stop at
the aldehyde. Brominating Br2 in water gives full
tri-bromination of phenol. Dehydration of a 2∘ alcohol
needs strong acid and heat. Reduction of a ketone to a
2∘ alcohol is done with NaBH4 or LiAlH4.
(i) 1∘ alcohol → acid. Reagents:
acidified KMnO4 (or alkaline KMnO4
followed by H3O+); also acidified
K2Cr2O7, or chromic acid (CrO3 in
H2SO4).
(ii) 1∘ alcohol → aldehyde.
Reagent: pyridinium chlorochromate (PCC) in
CH2Cl2. PCC is mild enough to stop at the
aldehyde without over-oxidising to acid.
(iii) Phenol → 2,4,6-tribromophenol.
Reagent: bromine water (Br2 in H2O). The
polar aqueous solvent supports the phenoxide form;
bromination proceeds three times consecutively at
each ortho/para position to give a white precipitate
of 2,4,6-tribromophenol.
(iv) Benzyl alcohol → benzoic acid.
Reagents: acidified KMnO4 (or
K2Cr2O7/H2SO4), or alkaline KMnO4
followed by acid workup.
(v) Propan-2-ol → propene. Reagent:
concentrated H2SO4 at 440K (alcohol
dehydration, E1 mechanism, see Q 7.19). Alternative:
H3PO4 or anhydrous Al2O3 at higher
temperature.
(vi) Butan-2-one → butan-2-ol. Reagent:
NaBH4 (sodium borohydride) in ethanol, or
LiAlH4 in dry ether. Both deliver a hydride to
the carbonyl carbon; aqueous workup gives the
2∘ alcohol.
(i) acidified KMnO4;
(ii) PCC in CH2Cl2;
(iii) Br2 in water;
(iv) acidified KMnO4;
(v) conc. H2SO4, 440K;
(vi) NaBH4 (or LiAlH4).
KV
Kavya Verma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Six different reagents, each
tied to a specific functional-group change. Memorise the
matchings rather than just the names.
(i) and (iv) both go from -CH2OH (or
-CHO) to -COOH; any strong oxidant works.
Acidified KMnO4 is the canonical choice.
(ii) needs to stop one oxidation level short. PCC is
the textbook reagent because it does not contain any
water (so it cannot hydrolyse the aldehyde further to
a hydrate then to an acid).
(iii) In water, the bromination goes all the way to
the 2,4,6-tribromide because each successive
bromination is still fast on the highly activated
ring; once the three positions are filled, the
substitution stops on its own.
(v) Concentrated H2SO4 at 440K is the
standard E1 dehydration condition; lower temperature
(413K) would have given diisopropyl ether
instead.
(vi) Carbonyl reduction. NaBH4 is selective for
ketones and aldehydes; it does not touch esters or
acids. LiAlH4 is a more aggressive hydride and
will reduce esters too.
Why this matters. Mastering the reagent-product
matching is the single most rewarding investment for the
chemistry portion of any entrance exam.
Reagent list as in the main solution.
Q 7.22
Give reason for the higher boiling point of ethanol in comparison to methoxymethane.
Concept used. Ethanol CH3-CH2-OH and
methoxymethane (dimethyl ether) CH3-O-CH3 have the
same molecular formula C2H6O and the same molecular
mass (46 g/mol), but very different boiling points: ethanol
78.4, dimethyl ether -24.8.
The gap of ∼100 is due to hydrogen bonding.
Ethanol has an -OH hydrogen: the H is bonded to
a highly electronegative O. So ethanol acts as a
hydrogen-bond donor in
R-O-H ⋯ O(H)-R.
Methoxymethane does not have any -OH
hydrogen. Its only hydrogens are on carbon, and C-H
is too non-polar to donate a hydrogen bond. The
oxygen has lone pairs (it can be an acceptor) but
there is no partner that can donate.
So ethanol forms intermolecular H-bonds; dimethyl
ether does not. To vapourise ethanol you must break
these bonds (each ∼20kJ/mol); to vapourise
dimethyl ether you only fight dipole-dipole and
dispersion forces.
Numerical comparison. Heat of vaporisation:
Δ Hvap(ethanol) ≈
38.6kJ/mol;
Δ Hvap(Me2O) ≈
21.5kJ/mol.
The ∼17 kJ/mol gap is exactly the order of
magnitude of one or two hydrogen bonds per molecule.
Ethanol's -OH forms intermolecular hydrogen
bonds; dimethyl ether cannot (no H bonded to O). So ethanol
boils about 100 above dimethyl ether despite
having the same molecular mass.
SR
Sneha Reddy
M.Sc Chemistry, IIT Kanpur
Verified Expert
Picture-first. Draw both molecules and look at where
the polar bonds are.
Ethanol's polar bond is O-H (Δχ = 1.24). The
H is positive enough (δ+ ≈ +0.4 e) to
attract another molecule's O lone pair. Result:
chains of H-bonded ethanol molecules in the liquid.
Methoxymethane's most polar bond is C-O
(Δχ = 0.89). The C-H bonds are nearly
non-polar, so the molecule has only weak
dipole-dipole and London dispersion interactions.
The energetic cost of vapourisation is therefore much
higher for ethanol: 38.6kJ/mol vs
21.5kJ/mol.
By the Trouton's rule estimate, b.p. ∝Δ Hvap: a 17 kJ/mol gap gives roughly
a 100 boiling-point gap.
Why this matters. The same logic explains why water
(H-O-H) boils much higher than hydrogen sulfide
(H-S-H) despite H2S being heavier: only water
forms H-bonds.
Hydrogen bonding makes ethanol's b.p.
78 vs methoxymethane's -25.
Q 7.23
Give IUPAC names of the following ethers:
(i) C2H5-O-CH2-CH(CH3)-CH3
(ii) CH3-O-CH2-CH2-Cl
(iii) p-O2N-C6H4-O-CH3
(iv) CH3-CH2-CH2-O-CH3
(v) cyclohexane bearing gem-dimethyl groups at one ring carbon and -OC2H5 at the opposite (1,4) ring carbon
(vi) C6H5-O-C2H5.
Concept used. Same rules as in Q 7.1 for ethers. Name
the smaller R-O- side as an alkoxy substituent
(``methoxy'', ``ethoxy'') and treat it as a prefix on the longer
parent. For aromatic ethers, benzene (or a longer parent) is the
ring; the alkoxy group sits on it. Numbering chooses the lowest
locant for the alkoxy group when other choices are tied.
(i)C2H5-O-CH2-CH(CH3)-CH3: longer side
is the 3-carbon chain -CH2-CH(CH3)-CH3
(isobutyl). Numbering from the O-end places the
-OC2H5 (ethoxy) at C-1, the methyl branch at C-2.
Name: 1-ethoxy-2-methylpropane.
(ii)CH3-O-CH2-CH2-Cl: parent is
2-chloroethane (Cl-CH2-CH2-); methoxy is the
substituent at C-1, Cl at C-2. Both locants tie; use
alphabetical order to break the tie. Name:
1-chloro-2-methoxyethane.
(iii)p-O2N-C6H4-O-CH3: parent is
benzene; substituents are methoxy at C-1 and nitro at
C-4. Name: 1-methoxy-4-nitrobenzene
(or p-nitroanisole).
(iv)CH3-CH2-CH2-O-CH3: parent is
propane; methoxy at C-1. Name:
1-methoxypropane.
(v) A cyclohexane ring with two methyls at one
carbon (the gem-dimethyl carbon) and an -OC2H5 at
the opposite (1,4) carbon. No suffix-priority group is
present, so numbering is chosen to give the lowest
locant set across all substituents. Placing the two
methyls at C-1 (gem) and the ethoxy at C-4 gives the
locant set 1,1,4; the reverse choice (ethoxy at
C-1, methyls at C-4) gives 1,4,4. Set 1,1,4
wins at the second locant (1 < 4). Name:
4-ethoxy-1,1-dimethylcyclohexane.
(vi)C6H5-O-C2H5: parent is benzene
with -OC2H5 as the substituent. Name:
ethoxybenzene (common: phenetole).
Structural observation. Every ether name is
``alkyl-oxy-parent'' or ``aryl-oxy
-parent''. Identify the longer chain or ring first.
For acyclic ethers (i, ii, iv) the longer C-chain
attached to O is the parent; the shorter is the
-OR substituent.
For aromatic ethers (iii, vi) the benzene ring is the
parent (because it is a 6-carbon ring, longer than any
-OR). The substituent on the ring is the alkoxy.
For cyclic ether (v) the cyclohexane ring is the
parent; the substituents are two methyls (gem) at C-1
and an ethoxy at C-4 (chosen to give the lower locant
set 1,1,4).
Numbering rules: principal group gets the lowest
locant; if ties exist, alphabetical order of
substituents breaks the tie.
Why this matters. You will rely on the same
``alkoxy-on-parent'' template throughout the chapter for
products of Williamson synthesis.
Names as listed in the main solution.
Q 7.24
Write the names of reagents and equations for the
preparation of the following ethers by Williamson's synthesis:
(i) 1-Propoxypropane (ii) Ethoxybenzene
(iii) 2-Methoxy-2-methylpropane (iv) 1-Methoxyethane.
Concept used.Williamson synthesis: alkoxide
R-O-Na+ + primary alkyl halide → ether + NaX. The
SN2 step requires that the alkyl halide be primary or
methyl. For ethers with a 3∘ alkyl group, the alkoxide
must come from the 3∘ alcohol and the halide must be
primary, never the other way around.
(i) 1-PropoxypropaneCH3CH2CH2-O-CH2CH2CH3
(di-n-propyl ether). Both halves are n-propyl, so
use sodium n-propoxide and n-propyl bromide:
CH3CH2CH2-ONa + Br-CH2CH2CH3 -> CH3CH2CH2-O-CH2CH2CH3 + NaBr.
(ii) EthoxybenzeneC6H5-O-C2H5. The
alkoxide must come from phenol (sodium phenoxide),
and the alkyl halide must be primary ethyl iodide or
bromide:
C6H5-ONa + C2H5-Br -> C6H5-O-C2H5 + NaBr.
Reverse choice (C2H5-ONa + C6H5-X) fails because
C6H5-X does not undergo SN2.
(iii) 2-Methoxy-2-methylpropane(CH3)3C-O-CH3. The 3∘ butyl group is on
the alkoxide side (from tert-butanol); the methyl
side is the halide:
(CH3)3C-ONa + CH3-I -> (CH3)3C-O-CH3 + NaI.
Reverse choice (CH3ONa + (CH3)3C-Br)
would mostly give isobutylene by E2, not the ether.
Strategic angle. The retrosynthetic question for any
Williamson is: ``which side becomes the alkoxide, and which
side becomes the halide?'' Answer: the alkoxide is the
sp3-O- with a phenyl, vinyl, 3∘ alkyl, or other
group that cannot undergo SN2; the halide is the primary or
methyl side that can undergo SN2.
For (i), both sides are n-propyl, so any assignment
works. The classical choice is sodium propoxide +
propyl bromide; the byproduct is NaBr.
For (ii), benzene cannot undergo SN2 (sp2
carbon, partial π-bond C-X). So phenol must be on
the alkoxide side; ethyl bromide is the alkyl halide.
For (iii), the tert-butyl group is on the alkoxide
side. Reagents: dry tert-butanol + Na to
give sodium tert-butoxide, then CH3I.
For (iv), the two ways are symmetric; pick whichever
starting materials are cheaper.
Why this matters. Williamson synthesis is by far the
most common ether-making reaction. Knowing which side becomes
the alkoxide saves you from the most common student mistake.
See main solution for the four equations.
Q 7.25
Illustrate with examples the limitations of
Williamson synthesis for the preparation of certain types of
ethers.
Concept used. The Williamson SN2 step has the
usual SN2 requirements: the alkyl halide must be primary
(or methyl); secondary halides give some SN2 and some E2;
tertiary halides give exclusively E2 (alkene). Vinyl and
aryl halides do not react at all by SN2. So Williamson
synthesis cannot make any ether whose halide side would be
2∘, 3∘, vinyl or aryl.
Limitation 1: 3∘ alkyl halides. If you
try to make tert-butyl methyl ether by reacting
CH3-ONa with (CH3)3C-Br, the 3∘
halide gives isobutylene by E2 instead of the ether:
(CH3)3C-Br + CH3O-Na+ -> (CH3)2C=CH2 + CH3OH + NaBr.
Fix: invert the roles. Use (CH3)3C-O-Na+ and
CH3-I instead.
Limitation 2: aryl (or vinyl) halides. Aryl
halides such as C6H5-Br cannot undergo SN2
(the sp2 carbon and the partial π-overlap of
C-Br with the ring block back-side attack). So
you cannot make C6H5-O-R by reacting
R-O-Na+ with C6H5-X. Instead, you
must use sodium phenoxide C6H5-O-Na+ and the
primary alkyl halide R-X.
Limitation 3: secondary halides give mixtures.
With a 2∘ halide, E2 competes substantially.
For instance, isopropyl bromide + sodium ethoxide
gives a mixture of ethyl isopropyl ether and propene:
(CH3)2CH-Br + C2H5O-Na+ -> (CH3)2CH-O-C2H5 + CH3-CH=CH2 + NaBr.
Yield of the desired ether is modest.
Limitation 4: bulky alkoxides. A very hindered
alkoxide such as tert-butoxide (CH3)3C-O-
attacks even primary halides poorly because the
alkoxide cannot reach the back side. Williamson
synthesis is best with small, primary alkoxides.
Williamson works cleanly only when the alkyl halide
side is primary (or methyl). It fails (or gives the wrong
product) for 3∘, vinyl, and aryl halides, and gives
mixtures for 2∘ halides.
AJ
Ankit Joshi
M.Tech Chemical Engineering, IIT Delhi
Verified Expert
Structural observation. The SN2 transition state
needs the nucleophile to approach the carbon from the side
opposite the leaving group. Anything that blocks that approach
breaks the reaction.
A tertiary carbon has three alkyl groups around it
plus the leaving group; the back side is fully
shielded. The molecule sheds the leaving group
unimolecularly (SN1) or, in the presence of a base,
loses a proton from a β-carbon (E2) instead.
An aryl (or vinyl) carbon is sp2. Its σ*
orbital is rotated by 90∘ relative to the
back-side direction, so an incoming nucleophile cannot
align with it.
Secondary carbons are intermediate: SN2 works but
E2 competes. The strong base R-O- pulls a
β-proton at almost the same rate it displaces
the halide.
Bulky alkoxides are themselves blocked from approach.
Even a primary halide reacts only slowly with
tert-butoxide, and instead acts as a strong base
to deprotonate any acidic site.
Why this matters. These limits explain why Williamson
synthesis is best taught as ``always use the primary side
for the halide''. They also motivate alternative ether
syntheses such as acid dehydration of alcohols, alkoxymercuration
of alkenes, or Mitsunobu reactions in modern labs.
Limitations: no Williamson with tertiary, vinyl, or
aryl halides; mixtures with secondary halides; sluggish with
bulky alkoxides.
Q 7.26
How is 1-propoxypropane synthesised from propan-1-ol?
Write mechanism of this reaction.
Concept used.Acid-catalysed dehydration of a
primary alcohol at moderate temperature
(413K, ∼140) and excess alcohol
gives the symmetric ether (here 1-propoxypropane,
CH3CH2CH2-O-CH2CH2CH3). The mechanism is
SN2 with the alcohol as nucleophile on a
protonated second molecule of alcohol. Higher temperature would
give the alkene (propene) by E1; lower temperature stops at
the alkyl hydrogen sulphate. So the temperature window for
ether formation is narrow.
Step 1: protonate one molecule of propan-1-ol. Conc.
H2SO4 protonates the OH to a much better
leaving group:
CH3CH2CH2-OH + H+ <=> CH3CH2CH2-OH2+.
Step 2: a second molecule of propan-1-ol attacks the
protonated first molecule by SN2 from the back side
of the leaving water:
!$CH3CH2CH2-OH2+ + HO-CH2CH2CH3 -> CH3CH2CH2-OH+(CH2CH2CH3) + H2O$.
(Mid-product: protonated 1-propoxypropane.)
Step 3: a third base (the conjugate base HSO4-,
water, or another alcohol) removes the extra proton:
CH3CH2CH2-OH+(CH2CH2CH3) <=> CH3CH2CH2-O-CH2CH2CH3 + H+.
The catalyst H+ is regenerated.
Net reaction:
!$2 CH3CH2CH2-OH H2SO4, 413 K CH3CH2CH2-O-CH2CH2CH3 + H2O$.
!%
[See diagram in the PDF version]
Two propan-1-ol molecules: one is protonated on O to make -OH2+ a leaving group; the second alcohol's O lone pair (violet curly arrow) attacks the protonated α-C by SN2, expelling water. Loss of H+ gives the neutral ether.
Dehydrate two molecules of propan-1-ol with conc.
H2SO4 at ∼413K. Mechanism: protonation →SN2 by a second alcohol on the protonated first →
deprotonation → ether.
PM
Pooja Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Picture-first. Think of one molecule as
``nucleophile'' (the un-protonated alcohol) and the other as
``electrophile'' (the protonated alcohol with -OH2+ as the
leaving group). The carbon between them is primary, so the
back-side displacement is easy.
At 413K, the equilibrium favours SN2 of
alcohol on protonated alcohol over E1 elimination.
Primary cations are unstable, so the unimolecular E1
pathway is suppressed.
The SN2 step happens twice in succession (each end
of the new C-O bond comes from a different alcohol
molecule), but is effectively one back-side attack
with H2O as leaving group.
The reaction is limited to symmetrical ethers between
primary alcohols. Trying to make (CH3)3C-O-CH3
by this method would not work (E1 elimination of
tert-butyl alcohol would dominate).
For unsymmetrical ethers, use Williamson synthesis
(Q 7.24).
Why this matters. This is the cheapest industrial
route to symmetric ethers like diethyl ether (anaesthetic) and
dipropyl ether (solvent).
14pt!2 CH3CH2CH2OH conc. H2SO4, 413 K CH3CH2CH2-O-CH2CH2CH3 + H2O. [4pt] Mechanism: SN2 of alcohol on protonated alcohol.
Q 7.27
Preparation of ethers by acid dehydration of
secondary or tertiary alcohols is not a suitable method. Give
reason.
Concept used. Whether acid dehydration of an alcohol
gives an ether (via SN2) or an alkene (via E1) depends on
how stable the carbocation intermediate is. Primary alcohols
have unstable 1∘ cations, so SN2 dominates (= ether).
2∘ and 3∘ alcohols have much more stable cations,
so E1 elimination dominates (= alkene). At the temperatures
needed for any ether-forming step, the elimination has already
taken over.
Recall the two competing pathways from a protonated
alcohol R-OH2+:
SN2 with a second alcohol gives an ether;
E1 (loss of β-H from the carbocation) gives
an alkene.
Carbocation stability: 3∘ > 2∘ > 1∘.
For a 3∘ alcohol, the cation R3C+ forms
easily; β-H abstraction (E1) is fast; alkene
is the major product. For a 2∘ alcohol, E1
is again dominant though somewhat slower.
Steric factor. The SN2 ether-forming step requires
another alcohol molecule to attack the carbon bearing
the leaving group. A 3∘ carbon is too crowded
for SN2 by any nucleophile; a 2∘ carbon is
modestly hindered. Both factors push the reaction
toward elimination.
Worked example. tert-butanol with conc.
H2SO4 at 413K gives isobutylene
((CH3)2C=CH2), not tert-butyl ether. The
ether route fails.
Conclusion. Use acid dehydration only for primary
alcohols (or for symmetric secondary ethers under
controlled conditions). For 3∘ or
unsymmetrical ethers, use Williamson synthesis
instead.
For 2∘ or 3∘ alcohols the protonated
intermediate undergoes E1 (loss of β-H from a stable
carbocation) much faster than SN2 by another alcohol, so
the alkene is the major product and very little ether forms.
AK
Arjun Kapoor
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Think of SN2 vs E1 as a race
between two pathways out of the same intermediate. Whichever
is faster wins; for 2∘ and 3∘ alcohols, E1 wins
by a wide margin.
Once a 3∘ alcohol is protonated, the leaving
water departs spontaneously to give a stable 3∘
cation. The cation is too short-lived (and the
approach to its sp2 carbon too crowded for any
second alcohol to make a back-side attack.
Instead, a nearby base (water, HSO4-, even
another alcohol acting as a base) plucks a
β-proton; the C-H electrons drop into the empty
p-orbital and the alkene is formed.
For a 2∘ alcohol, the same logic applies but
less extreme: ∼80% alkene, ∼20% ether at
best.
The ``no good'' verdict in NCERT therefore means
2∘ alcohols give a mixture (with the alkene
dominating) and 3∘ alcohols give essentially
100% alkene. Use Williamson instead.
Why this matters. This is one of those important
``don't make this product this way'' lessons. Knowing why a
method fails is as useful as knowing why one succeeds.
Acid dehydration of 2∘ or 3∘ alcohols
gives mainly the alkene via E1, not the ether. Use
Williamson instead.
Q 7.28
Write the equation of the reaction of hydrogen iodide
with:
(i) 1-propoxypropane (ii) methoxybenzene
(iii) benzyl ethyl ether.
Concept used.Cleavage of ethers by HI
proceeds in two main ways: (a) with an alkyl-alkyl ether, the
smaller alkyl group goes to iodine and the larger one
retains the OH (via SN2); (b) with an alkyl-aryl ether,
the aryl-O bond is unbreakable, so the alkyl group goes to
iodine and the aryl group retains the OH (giving phenol).
With excess HI, both halves of an alkyl-alkyl ether become
alkyl iodides. The reaction is SN2 in cold HI for primary
alkyl groups and SN1 in hot HI for tertiary alkyl groups.
(i) 1-Propoxypropane + HI. Both alkyl groups
are n-propyl. Mechanism: protonate the ether O,
then I- attacks one of the C atoms by SN2,
breaking the C-O bond. With one equivalent of HI:
CH3CH2CH2-O-CH2CH2CH3 + HI -> CH3CH2CH2-I + CH3CH2CH2-OH.
With excess HI, the alcohol product reacts further:
CH3CH2CH2-OH + HI -> CH3CH2CH2-I + H2O.
(ii) Methoxybenzene (anisole) + HI. Anisole
is C6H5-O-CH3. The aryl-O bond is too strong
(and aryl carbon does not undergo SN2). So
I- attacks the methyl carbon, giving methyl
iodide and phenol:
C6H5-O-CH3 + HI -> C6H5-OH + CH3-I.
Note that the phenol does not react further
with HI to give an aryl iodide (aryl C-O is stable).
(iii) Benzyl ethyl ether + HI.C6H5-CH2-O-C2H5. The benzyl carbon is
special: it forms a very stable benzyl cation. Under
acidic conditions, the cleavage goes by SN1 at
the benzyl side. Iodide attacks the benzyl cation,
giving benzyl iodide and ethanol:
C6H5-CH2-O-C2H5 + HI -> C6H5-CH2-I + C2H5-OH.
(i) C3H7-O-C3H7 + HI -> C3H7-I + C3H7-OH;
(ii) C6H5-O-CH3 + HI -> C6H5-OH + CH3-I;
(iii) C6H5-CH2-O-C2H5 + HI -> C6H5-CH2-I + C2H5-OH.
RI
Rahul Iyer
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. For every ether cleavage by HI,
identify the weakest C-O bond (the one that breaks) and
follow it: the carbon on that side picks up the iodide,
and the carbon on the other side keeps the oxygen (as OH).
In (i), both carbons are primary alkyl; either C-O
breaks with equal probability. By stoichiometry,
one mole of HI cleaves only one C-O bond,
giving 1 mole of propyl iodide and 1 mole of
propan-1-ol.
In (ii), the aryl-O bond is too strong to cleave
(π-conjugation locks it in place). HI must
attack the methyl carbon by SN2, displacing
C6H5-O^- which then picks up a proton.
In (iii), the benzyl carbon stabilises a positive
charge through resonance with the ring. So under
the acidic conditions HI provides, an SN1 cleavage
is possible at the benzyl C. Iodide traps the cation.
The ethyl group keeps the O (as ethanol).
All three reactions are exothermic and quantitative
in laboratory practice.
Why this matters. The cleavage rule
``aryl side keeps the OH; benzyl side becomes the iodide''
is a high-yield JEE question and a useful synthetic tool for
hydrolysing methyl protective groups on phenols.
Explain the fact that in aryl alkyl ethers (i) the
alkoxy group activates the benzene ring towards electrophilic
substitution and (ii) it directs the incoming substituents to
ortho and para positions in the benzene ring.
Concept used. Same logic as Q 7.16. The
alkoxy group -OR has a lone pair on oxygen that
can donate π-density into the ring (+M, mesomeric
donation). The ring becomes electron-rich (activation), and
the donation pushes excess density to the ortho and para
positions (directing).
Activation: lone pair pushes density into the
ring. Draw resonance structures for the aryl alkyl
ether Ar-O-R. The lone pair on O donates into
the ring, putting δ- on the ortho and para
carbons:
Ar-O+R <=> -Ar=O+R (with the negative
charge on the ring's o/p carbons).
So the ring is more nucleophilic than benzene
itself; an electrophile attacks faster.
Directing to o/p: stable arenium intermediate
at o/p. When E+ attacks the ortho or para
carbon, the resulting arenium ion has a resonance
structure with the positive charge directly on the
carbon bonded to OR. The OR oxygen's lone pair
donates into that empty orbital, forming an
oxocarbenium-like resonance structure with all
atoms having full octets. This contributes a large
stabilisation.
For meta attack, the positive charge lands on
carbons not bonded to OR, so the oxygen lone pair
cannot help. The meta intermediate is therefore much
less stable than the o/p intermediates.
Experimental confirmation: anisole reacts with
Br2 in glacial acetic acid at room temperature
to give about 90% p-bromoanisole and 10%
o-bromoanisole, with no meta isomer at all.
(i) Activation: -OR donates π-density into
the ring through O's lone pair (+M), raising the ring's
HOMO and stabilising every transition state of EAS.
(ii) o/p directing: the arenium intermediate at o/p has an
oxocarbenium resonance structure (full-octet), absent for
meta. So o/p attack is much more favourable.
PD
Pranav Desai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Two questions reduce to one
observation: the lone pair on O delocalises into the ring,
both in the ground state (activation) and in the EAS
transition state at o/p (directing).
Sketch the ground state. In C6H5-OR, oxygen has
two lone pairs; one is in a p-orbital aligned with
the ring's π-system. That lone pair overlaps and
donates π-density.
Sketch the arenium intermediate for o-attack: the
+ charge sits on C-2 (alongside the OR oxygen).
Oxygen pushes its lone pair into the cation, forming
a Friedel-Crafts-like ``oxocarbenium'' resonance form.
Same drawing for p-attack: the positive charge again
ends up on the OR-bearing C-1; oxygen's lone pair
stabilises it the same way.
For m-attack, the + charge sits on C-3 or C-5; the
OR oxygen at C-1 cannot reach them with its lone
pair. So the meta arenium ion is much less stabilised.
Why this matters. Predicting o/p vs m directing is
the bread-and-butter of EAS problems. The same logic applies
to -OH, -OR, -NH2, -NR2.
The -OR lone pair pushes π-density into the
ring (activation) and stabilises only the o and p arenium
intermediates (o/p directing).
Q 7.30
Write the mechanism of the reaction of HI with
methoxymethane.
Concept used. Methoxymethane (dimethyl ether),
CH3-O-CH3, reacts with HI by an SN2 mechanism. Step 1
protonates the ether oxygen, turning it into a leaving group;
step 2 sees I- attack one of the methyl carbons from
the back side, displacing methanol; step 3 (if excess HI is
present) protonates and substitutes the methanol to give a
second molecule of methyl iodide.
Step 1: protonation. The ether oxygen is mildly basic;
in HI, one of its lone pairs picks up a proton:
CH3-O-CH3 + HI <=> CH3-O(+)(H)-CH3 + I-.
The protonated ether (an oxonium ion) now has a much
better leaving group: CH3OH.
Step 2: SN2 by iodide. The iodide ion attacks one
methyl carbon from the back side of the leaving
O(H)CH3 group; the C-O bond breaks as the new
C-I bond forms:
I- + CH3-O(+)(H)-CH3 -> CH3-I + HO-CH3.
The first methyl carbon is now in CH3-I; the
other methyl carbon stays in CH3-OH (methanol).
Step 3: with excess HI, the methanol also reacts. It
is first protonated to CH3-OH2+, then
iodide displaces water by SN2:
CH3OH + HI -> CH3-I + H2O.
Net product with excess HI: 2 equiv of CH3I
and 1 equiv of H2O.
!%
[See diagram in the PDF version]
Curly arrows: (1) ether O lone pair grabs H+ of HI; (2) I- attacks one CH3 from the back side (SN2), breaking the C-O bond and expelling CH3OH.
Mechanism: (1) HI protonates the ether oxygen to give
an oxonium ion; (2) I- attacks a methyl carbon by
SN2, displacing CH3OH; (3) excess HI converts the
methanol to a second equivalent of methyl iodide.
AS
Aanya Sharma
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. The SN2 step is the
rate-determining step. The reaction is fastest with the most
nucleophilic halide ion among HF, HCl, HBr, HI. Iodide
is the largest, softest, and most nucleophilic anion in the
series; that is why HI works so well for ether cleavage.
Both methyl carbons are equivalent in dimethyl ether,
so iodide can attack either: the rate is twice that
of attack on a single methyl.
The SN2 transition state has trigonal-bipyramidal
geometry around the attacked carbon, with I-
and the leaving methanol on opposite sides.
In water-free conditions, the released methanol is
protonated again by HI and converted to CH3I,
so the final yield is 2 mol CH3I per mol of
ether (with 1 mol of water as the only other
product).
The reaction works for any methyl-methyl, methyl-primary,
or primary-primary ether by the same mechanism. For
tert-alkyl ethers, SN1 takes over.
Why this matters. The reaction is the
``demethylation'' workhorse: protecting an OH as an
OMe ether and then removing the methyl with HI lets a chemist
work on the rest of the molecule unhindered.
Mechanism: protonation →SN2 by I- at
the methyl carbon →CH3I + CH3OH (and with excess
HI, a second equivalent of CH3I from the methanol).
Q 7.31
Write equations of the following reactions:
(i) Friedel-Crafts reaction - alkylation of anisole.
(ii) Nitration of anisole.
(iii) Bromination of anisole in ethanoic acid medium.
(iv) Friedel-Craft's acetylation of anisole.
Concept used. Anisole is C6H5-OCH3
(methoxybenzene). The -OCH3 group is a strong +M ring
activator and an ortho/para director (Q 7.29). So every
electrophilic aromatic substitution on anisole goes mainly
para (and some ortho), with para usually the major isomer due
to lower steric clash with -OCH3.
(i) Friedel-Crafts alkylation. React anisole
with an alkyl halide R-Cl in the presence of
anhydrous AlCl3. The Lewis acid ionises the
alkyl halide to a carbocation R+, which
attacks the activated ring. With CH3Cl:
C6H5-OCH3 + CH3-Cl AlCl3 p-CH3O-C6H4-CH3 + HCl
(major) plus some o-methylanisole.
(ii) Nitration of anisole. Mix anisole with
a 1:1 mixture of conc. HNO3 and conc.
H2SO4 at 20; mostly the
para-nitro product forms (with some
ortho):
C6H5-OCH3 + HNO3 H2SO4 p-CH3O-C6H4-NO2 + H2O
(major) plus some o-nitroanisole.
(iii) Bromination in ethanoic acid. Anisole
+ Br2 in glacial CH3COOH at
0 gives mainly p-bromoanisole
(yield about 90%) and a small amount of
o-bromoanisole:
C6H5-OCH3 + Br2 CH3COOH p-CH3O-C6H4-Br + HBr.
(iv) Friedel-Crafts acetylation. React
anisole with acetyl chloride CH3COCl in the
presence of anhydrous AlCl3 in CS2:
C6H5-OCH3 + CH3-CO-Cl AlCl3 p-CH3O-C6H4-CO-CH3 + HCl.
The major product is para-methoxy
acetophenone.
0.95!%
[See diagram in the PDF version]
The -OCH3 group is at C-1; the electrophile (CH3, NO2, Br, COCH3) lands predominantly at the para (C-4) position. A small amount of the corresponding ortho isomer also forms.
All four reactions give predominantly the
para isomer because the -OCH3 group is a
strong o/p director and the para position is sterically
preferred over ortho.
VJ
Vivaan Joshi
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Anisole is the textbook activated
arene for Friedel-Crafts and nitration/bromination. Predict
the product by drawing the o/p resonance stabilisation of
the arenium ion.
In each of the four reactions, generate the
electrophile first: alkylation
(R+ from R-Cl/AlCl3), nitration
(NO2+ from HNO3/H2SO4), bromination
(Br+ from Br2 in CH3COOH),
acetylation (CH3CO+ from
CH3COCl/AlCl3).
Each electrophile attacks the ring at the o or p
position; para is sterically preferred.
The leaving group from the arenium ion is H+,
which gets sucked up by Cl- (or HSO4-,
etc.) to give HCl (or H2SO4).
In aqueous-free conditions (e.g., for the
Friedel-Crafts), the catalyst AlCl3 is
regenerated and continues to ionise more electrophile.
Why this matters. The reactivity of an activated
arene like anisole is the basis of dye chemistry,
pharmaceutical synthesis, and the production of vanillin and
eugenol.
Equations as written; in each case the major
product is the para isomer.
Q 7.32
Show how would you synthesise the following alcohols
from appropriate alkenes?
(i) 1-methylcyclohexan-1-ol
(ii) 4-methylheptan-4-ol
(iii) pentan-2-ol
(iv) 2-cyclohexylbutan-2-ol.
Concept used. To make a tertiary or secondary alcohol
from an alkene we use Markovnikov hydration (acid +
water, or oxymercuration/demercuration), placing OH on the
more substituted carbon. For each target alcohol, identify
the carbon that carries OH and work backwards to the alkene
formed by removing OH and a β-H.
(i) 1-methylcyclohexan-1-ol (3∘
alcohol on a cyclohexane ring with a methyl at the
same carbon). Start from 1-methylcyclohex-1-ene:
1-methylcyclohex-1-ene + H2O H2SO4 1-methylcyclohexan-1-ol.
Markovnikov adds OH to the more substituted (and
3∘) carbon.
(ii) 4-methylheptan-4-ol (a 3∘
alcohol with a methyl, an n-propyl and an n-propyl
group all on the OH-bearing C-4 of heptane). Start
from 4-methylhept-3-ene. Markovnikov hydration puts
OH on the more substituted carbon (the C bearing the
methyl), giving the 3∘ alcohol directly:
4-methylhept-3-ene + H2O H2SO4 4-methylheptan-4-ol.
(iii) Pentan-2-ol (a 2∘ alcohol,
CH3-CH(OH)-CH2-CH2-CH3). Hydrate pent-1-ene
(CH2=CH-CH2-CH2-CH3) under Markovnikov
conditions; OH lands on the more substituted internal
carbon (C-2):
CH2=CH-CH2-CH2-CH3 + H2O H2SO4 CH3-CH(OH)-CH2-CH2-CH3.
(iv) 2-cyclohexylbutan-2-ol.
Structure: C6H11-C(OH)(CH3)-CH2-CH3. Hydrate
2-cyclohexylbut-2-ene
(C6H11-C(CH3)=CH-CH3); OH lands on the more
substituted (cyclohexyl-bearing) carbon, giving the
3∘ alcohol:
C6H11-C(CH3)=CH-CH3 + H2O H2SO4 C6H11-C(OH)(CH3)-CH2-CH3.
Each alcohol is made by Markovnikov hydration of
the alkene that results from removing OH + a β-H from
the target. Reagent in each case: dil. H2SO4 +
H2O (or oxymercuration-demercuration).
KP
Karan Pillai
M.Sc Chemistry, IIT Kanpur
Verified Expert
Strategic angle. Mark the OH-bearing carbon and a
neighbouring carbon with one fewer H atoms; that bond is the
double bond of the precursor alkene. Markovnikov regiochemistry
guarantees the same regio outcome on hydration.
For (i), the 3∘ OH is on C-1 of cyclohexane,
with a methyl also at C-1. Removing OH from C-1 and
a H from C-2 gives the disubstituted endocyclic
alkene 1-methylcyclohex-1-ene. Markovnikov adds
H-OH back: OH lands at C-1 again, as required.
For (ii), the 3∘ OH at C-4 of heptane bears a
methyl branch; removing OH and a β-H from C-3
gives 4-methylhept-3-ene. Markovnikov hydration
replaces the OH cleanly on the more substituted
(methyl-bearing) carbon.
For (iii), pentan-2-ol is made from pent-1-ene by
Markovnikov hydration; OH lands on the more
substituted internal C-2.
For (iv), the alkene is 2-cyclohexylbut-2-ene;
Markovnikov adds OH to the more substituted
(cyclohexyl-and-methyl-bearing) carbon, giving the
3∘ alcohol 2-cyclohexylbutan-2-ol.
Why this matters. Retrosynthetic disconnection of an
alcohol to an alkene is a standard exam exercise; the same
two carbons that flank the original C=C bond now flank the
new C-O bond.
Reagents: dil H2SO4 + H2O in each
case; alkene precursors as listed in the main solution.
Q 7.33
When 3-methylbutan-2-ol is treated with HBr, the
following reaction takes place: CH3-CH(CH3)-CH(OH)-CH3 + HBr -> CH3-CBr(CH3)-CH2-CH3.
Give a mechanism for this reaction.
(Hint: the 2∘ carbocation formed in step II rearranges
to a more stable 3∘ carbocation by a hydride ion shift
from the 3rd carbon atom.)
Concept used.Acid-catalysed conversion of an
alcohol to an alkyl halide via SN1: protonation of OH,
loss of water to give a carbocation, and capture of the cation
by the halide. If the initially formed cation can rearrange
by a hydride shift or methyl shift to a
more stable cation, it does so before being captured. Here
the secondary cation rearranges to a tertiary cation.
Step 1: protonation. HBr donates a proton to
the OH of 3-methylbutan-2-ol:
!$CH3-CH(CH3)-CH(OH)-CH3 + HBr <=> CH3-CH(CH3)-CH(OH2+)-CH3 + Br-$.
The OH is now an excellent leaving group (H2O).
Step 2: loss of water (slow, R-D step). The
protonated alcohol loses water unimolecularly to give
a secondary carbocation at C-2:
CH3-CH(CH3)-CH(OH2+)-CH3 -> CH3-CH(CH3)-CH(+)-CH3 + H2O.
Call this cation A (the 2∘ cation
at C-2).
Step 3: 1,2-hydride shift. The hydrogen on
C-3 (the carbon adjacent to the cation, which bears a
methyl group) migrates with its bonding electron pair
to C-2. The positive charge moves from C-2 to C-3,
where it sits on a now-tertiary carbon:
CH3-CH(CH3)-CH(+)-CH3 -> CH3-C(+)(CH3)-CH2-CH3.
Call this cation B (the more stable 3∘
cation at C-3). The shift is essentially barrierless
because it gives a much more stable cation.
Step 4: capture by bromide. The bromide ion
attacks the 3∘ cation from either face,
giving the product:
CH3-C(+)(CH3)-CH2-CH3 + Br- -> CH3-CBr(CH3)-CH2-CH3.
Final product: 2-bromo-2-methylbutane.
!%
[See diagram in the PDF version]
The violet curly arrow shows the C-H bonding pair migrating from C-3 to C-2 (a 1,2-hydride shift), promoting the 2∘ cation to a 3∘ cation before Br- traps it.
Mechanism: protonation of OH → loss of water to
give a 2∘ cation → 1,2-hydride shift from C-3 to
C-2, producing a 3∘ cation → capture by Br-
to give 2-bromo-2-methylbutane.
AM
Aarav Mehta
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Strategic angle. Whenever the obvious carbocation
intermediate has a neighbouring carbon that would, on a 1,2
shift, give a more substituted cation, the rearrangement
happens. Here the obvious cation is 2∘ at C-2;
the C-3 carbon already carries a methyl branch, so a hydride
shift from C-3 promotes the cation to 3∘.
Protonation of the OH is the standard first step in
any acid-catalysed alcohol substitution. The
protonated form has a very weak C-OH2+ bond.
The C-OH2+ bond breaks heterolytically, releasing
water and leaving a 2∘ cation at C-2. This is
the slow step.
Within nanoseconds, the cation undergoes a 1,2-H shift
from C-3. The migrating hydride brings its bonding
electrons with it; the positive charge moves to C-3,
which now has three C substituents.
The tertiary cation is trapped by bromide, giving
2-bromo-2-methylbutane. Note that no Br is on
C-2 (the original OH carbon); rearrangement has
moved the substitution one carbon over.
Why this matters. Carbocation rearrangements are a
classic ``why doesn't the obvious product form'' question.
Always check for a neighbouring carbon that, after a 1,2
shift, gives a more stable cation.
Mechanism: protonation → loss of H2O to
2∘ cation → 1,2-hydride shift to 3∘ cation
→ trap by Br-. Product: 2-bromo-2-methylbutane.
NCERT Solutions for Class 12 Chemistry: All Chapters
Also Check: CBSE Class 12 Chemistry Syllabus 2026-27
NCERT Solutions for Class 12 Chemistry Chapter 7 - FAQs
Q1. How many questions are there in NCERT Class 12 Chemistry Chapter 7 Alcohols, Phenols and Ethers exercise?
The main exercise of Chapter 7 contains 32 questions, supplemented by 12 intext questions distributed across the chapter. All 44 questions are solved step-by-step in the Collegedunia PDF on this page.
Q2. What is the most important named reaction from Chapter 7 Alcohols, Phenols and Ethers for CBSE 2026?
Kolbe's reaction (sodium phenoxide + CO2 -> salicylic acid) and Reimer-Tiemann reaction (phenol + CHCl3 / NaOH -> salicylaldehyde) are the two highest-yield named reactions, appearing in CBSE 2023, 2024, and 2025 in different forms.
Q3. Why is phenol more acidic than ethanol?
Phenoxide ion (the conjugate base of phenol) is stabilised by resonance over the aromatic ring, with the negative charge delocalised onto the ortho and para positions. Ethoxide has no such delocalisation, so phenol releases the proton more easily.
Q4. What is the difference between Williamson synthesis and Markovnikov hydration?
Williamson synthesis forms an ether by SN2 attack of an alkoxide on a primary alkyl halide. Markovnikov hydration adds water across an alkene with the OH on the more-substituted carbon. Both are core preparation methods in Chapter 7 but for different functional groups.
Q5. Is Chapter 7 Alcohols, Phenols and Ethers part of the 2026-27 syllabus?
Yes, the chapter is fully retained in the current NCERT print and contributes 6-8 marks to the CBSE Class 12 Chemistry theory paper. No sub-topics from this chapter have been trimmed in the latest edition.
Q6. How do I use the Lucas test to distinguish primary, secondary, and tertiary alcohols?
Mix the alcohol with Lucas reagent (concentrated HCl + ZnCl2). Tertiary alcohols give immediate turbidity, secondary alcohols give turbidity in 5-10 minutes, and primary alcohols show no turbidity at room temperature. The Collegedunia solutions PDF includes a one-page Lucas-test summary chart.
Q7. What is the Williamson ether synthesis, and which alkyl halide should you use?
Williamson ether synthesis is the SN2 reaction of a sodium alkoxide (R-O-Na+) with an alkyl halide (R'-X) to give the ether R-O-R'. The alkyl halide MUST be primary; secondary and tertiary halides undergo E2 elimination with the strongly basic alkoxide and give alkenes instead. For an unsymmetrical ether, always derive the alkoxide from the bulkier alkyl group and the halide from the smaller, primary alkyl group.
Q8. What is the cumene process for preparing phenol, and what is the by-product?
The cumene process is the major industrial route to phenol. Cumene (isopropylbenzene) is oxidised by atmospheric O2 to cumene hydroperoxide, which on treatment with dilute sulphuric acid rearranges to phenol and acetone. Acetone is the valuable co-product, which makes the route economically attractive. The reaction is examinable in CBSE 5-mark questions; always name acetone as the co-product to score full marks.
Q9. How is salicylic acid prepared from phenol (Kolbe reaction)?
Sodium phenoxide is heated with CO2 at 400 K and 4 to 7 atm; the carboxylate intermediate is acidified to give salicylic acid (2-hydroxybenzoic acid). The mechanism involves electrophilic attack of CO2 on the activated ortho carbon of the phenoxide. The Kolbe reaction is the industrial route to salicylic acid, the precursor of aspirin. CBSE 2024 and 2025 both asked the full mechanism for 3 marks.
Q10. What is the difference between Markovnikov hydration and hydroboration-oxidation for preparing alcohols from alkenes?
Markovnikov hydration uses dilute H2SO4 and gives the Markovnikov alcohol (OH on the more-substituted carbon) via a carbocation intermediate; rearrangement is possible. Hydroboration-oxidation uses B2H6 in THF followed by alkaline H2O2 and gives the anti-Markovnikov alcohol (OH on the less-substituted carbon) via a concerted syn-addition. Hydroboration is rearrangement-free, which is why CBSE prefers it in 3-mark synthesis questions.
Q11. How is picric acid (2,4,6-trinitrophenol) prepared from phenol?
Picric acid is prepared by stepwise nitration of phenol. Treatment with dilute HNO3 at low temperature gives ortho- and para-nitrophenol; further nitration with more concentrated HNO3 gives 2,4-dinitrophenol; and final nitration with a mixture of concentrated HNO3 and H2SO4 gives picric acid. Picric acid has pKa 0.4 and is stronger than acetic acid, because three -NO2 groups stabilise the conjugate base by resonance and -I effects.
Q12. Why does anisole react with HI to give phenol and methyl iodide, not iodobenzene and methanol?
In anisole (C6H5-O-CH3), the phenyl-oxygen bond has partial double-bond character because the oxygen lone pair conjugates with the aromatic ring. This makes the aryl-O bond too strong to cleave. Instead, I- attacks the methyl carbon via SN2 at the sp3 centre, giving phenol (C6H5-OH) and methyl iodide (CH3-I). The same logic applies to all alkyl aryl ethers: cleavage always occurs at the sp3 alkyl carbon, never at the sp2 aryl carbon.
Q13. What is the Saytzeff rule for the acid-catalysed dehydration of alcohols?
Saytzeff's rule states that in an E1 dehydration of an alcohol, the more-substituted alkene (the more stable one) is the major product. For 2-methylbutan-2-ol with conc. H2SO4 at 443 K, the major product is 2-methylbut-2-ene (trisubstituted), not 2-methylbut-1-ene (disubstituted). The stability of the alkene is governed by hyperconjugation and is the controlling factor in E1 product distribution.
Q14. Where can I download the free PDF of NCERT Solutions for Class 12 Chemistry Chapter 7?
The free PDF is downloadable from the red button at the top of this page. The file is mobile-friendly, watermarked with the 2026-27 syllabus tag, and includes both the main exercise and intext-question solutions along with full step-by-step working for every named reaction (Lucas, Williamson, Reimer-Tiemann, Kolbe, cumene, Dow), acidity-order comparison, and mechanism walkthrough.














































































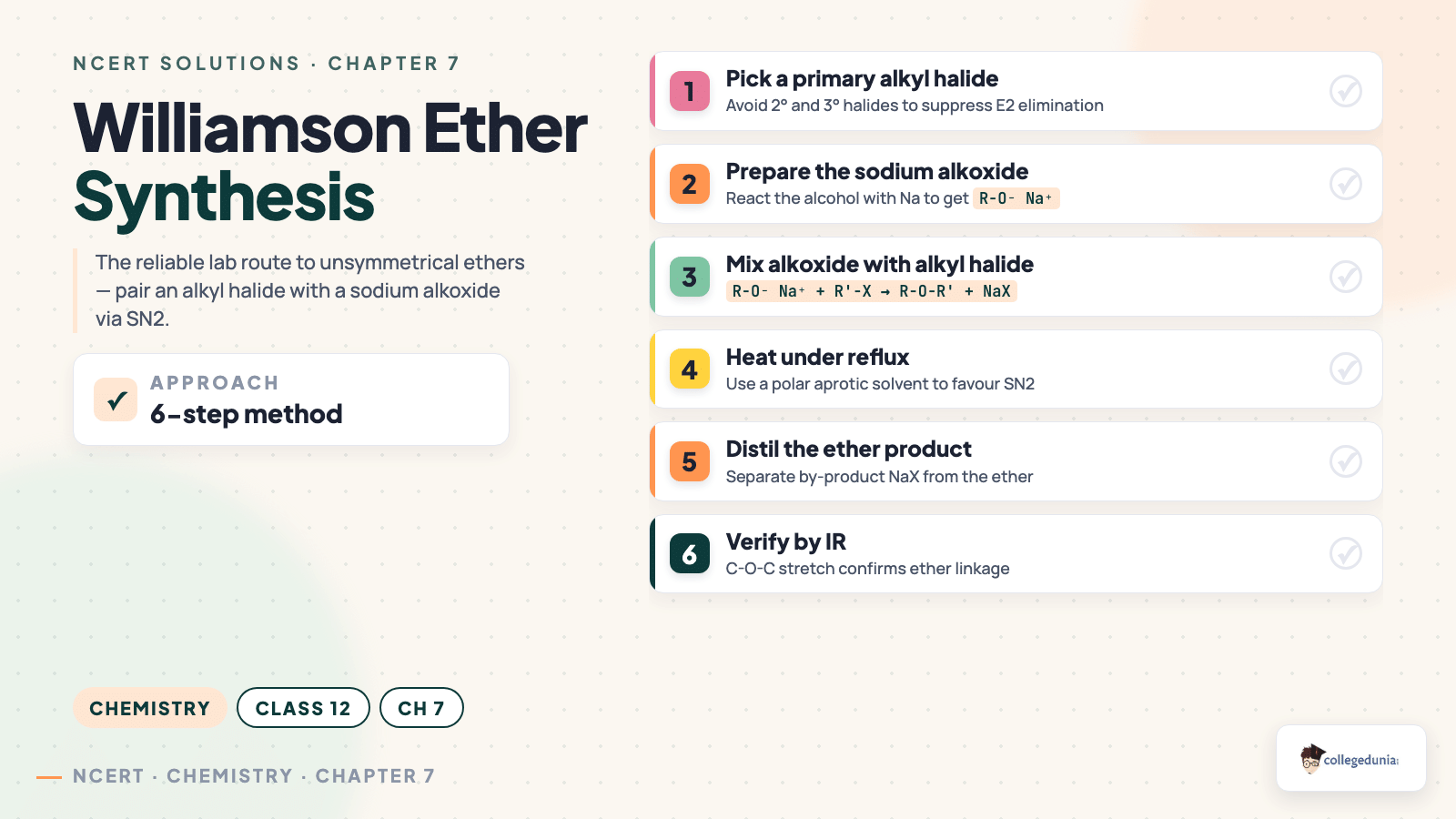



Comments