Senior Chemistry Editor | M.Sc. Chemistry, 12 Years | Updated on - May 25, 2026
Biomolecules are the organic compounds of life: carbohydrates, proteins, nucleic acids, vitamins and enzymes that run every cellular process. Class 12 Chemistry Chapter 10 Biomolecules sits at the close of the 2026-27 NCERT syllabus and is the cleanest bridge between Chemistry and the NEET Biology paper. This page hosts the worked Exemplar Solutions PDF.
CBSE Weightage: 3 to 5 marks (a 2-mark VSA on a biomolecule or deficiency disease, plus a 3-mark SA on protein structure, glycosidic linkage or DNA vs RNA)
JEE Main Weightage: 1 to 2% (~1 question per shift on carbohydrate classification, anomers, vitamins or nucleic-acid bases)
NEET Weightage: 1 to 2 questions per year on vitamins, enzyme classification and DNA/RNA bases; the chapter overlaps Class 12 Biology Unit 9
Each item is solved twice: a Solution gives the working, then an Expert's Solution names the controlling concept that decides the answer.
These Exemplar Solutions are curated by Collegedunia subject experts, mapped to the 2026-27 NCERT, and benchmarked against five years of CBSE, JEE Main and NEET papers.
Why the Biomolecules Exemplar Still Matters in the 2026-27 Syllabus
Biomolecules carries the lowest CBSE marks band in Class 12 Chemistry, yet it is the highest-yield chapter for NEET aspirants because every Exemplar fact reappears in the Biology paper.
NEET cross-paper overlap: Vitamin deficiency diseases, enzyme classes, DNA vs RNA bases and protein secondary structure are tested in both NEET Chemistry and NEET Biology - one revision earns marks twice.
Low-effort, high-recall: Most Exemplar items are recognition or classification, not multi-step synthesis. A focused 90-minute pass can lift the Organic block score by 3 to 5 marks on the Board paper.
Rationalisation impact: The 2026-27 NCERT keeps every biomolecule topic intact but moves the chapter from its older Chapter 14 slot to Chapter 10; "Hormones" was trimmed, so hormone-flavoured Exemplar items should be skipped.
How Collegedunia's Biomolecules Exemplar Solutions Help You Lock in the Marks
The Chapter 10 Exemplar rewards students who name the structural or functional reason a biomolecule behaves a certain way.
Every Question Type Worked End-to-End: MCQ-I, MCQ-II, SA, Matching and Assertion-Reason / LA, each with full reasoning.
Concept Stack Named: anomers, glycosidic linkages, protein 1° to 4° hierarchy, α-helix H-bonding, DNA vs RNA bases, 5′ to 3′ phosphodiester linkage, vitamin solubility, six enzyme classes.
NEET Bridge Tagged: Items that map onto NEET Biology Unit 9 are flagged so you score in both papers from one revision.
Biomolecules Exemplar: Question-Type Mix at a Glance
Chapter 10 splits into five question buckets. The mix below lets you triage between a one-sitting attempt and a two-day plan.
Question Type
Item Range
Count
Typical Marks (Board)
MCQ-I (single correct)
10.1 to 10.8
8
1
MCQ-II (multiple correct)
10.9 to 10.12
4
2
Short Answer (SA)
10.13 to 10.20
8
2 to 3
Matching Type
10.21
1
4
Assertion-Reason / LA
10.22 to 10.25
4
3 to 5
The 12 MCQ items together carry the entire recognition bucket: classification, vitamin identity, DNA bases and protein stabilisers.
Biomolecules Exemplar Step-Up from the NCERT Textbook
The Exemplar reframes textbook facts as multi-factor identification puzzles. Three concrete jumps:
Skill
NCERT Textbook Asks
Exemplar Asks
Polysaccharide structure
State glycogen is animal storage
Compare glycogen, amylose, amylopectin, cellulose on linkage (α-1,4 vs β-1,4) and branching (α-1,6); pick the branched pair
Protein structure
Define 1° to 4° structures
From a bond list (peptide, H-bonds, disulphide, van der Waals), pick stabilisers of α-helix vs fibrous proteins; cite the N−H···O=C rule
D/L vs (+)/(-) labels
Identify glucose as D-(+)
Decide if D-fructose is (+) or (-); explain why D cannot predict rotation
The shift is from single-fact recall to multi-factor classification. Every Expert's Solution names the controlling rule.
Biomolecules Class 12 Chemistry: Sample MCQ-II Solved with Branching-Pattern Walk-Through
MCQ-II is the highest-failure type because students stop after one correct option. The branching-polymer item is the canonical example.
Q (Exemplar 10.10): Which of the following carbohydrates are branched polymers of glucose?
(i) Amylose (ii) Amylopectin (iii) Cellulose (iv) Glycogen
Answer: (ii) Amylopectin and (iv) Glycogen.
Expert's reasoning: A branched glucose polymer needs both a chain linkage and a branch linkage: α-1,4 for the chain and α-1,6 for the branch. Amylose (only α-1,4) and cellulose (only β-1,4) are linear, so ruled out. Amylopectin carries both linkages with branches every ~25 residues; glycogen has the same dual linkages with branches every ~10 residues. So (ii) and (iv). Students who stop at one option lose the second mark - the MCQ-II rule is to check every option against the defining feature.
"α-1,4 + α-1,6 = branched, one linkage = linear" clears the polysaccharide bucket.
Carbohydrate Stereochemistry in the Exemplar: Anomers, Epimers and Mutarotation
Roughly a quarter of the MCQ-I and SA bucket on Chapter 10 rests on three closely-related stereochemistry terms. The Exemplar reframes them as multi-factor identification puzzles, not single-fact recall.
Anomers differ at the anomeric carbon (C1 in aldoses, C2 in ketoses). Alpha- and beta-D-glucopyranose are the canonical pair. Exemplar item 10.7 asks "which of the following are anomers" with a six-option grid; the lock-in rule is "only at the anomeric C, everything else identical".
Epimers differ at one non-anomeric chiral C. Glucose / galactose at C4, glucose / mannose at C2. Confusing anomers with epimers costs a 1- to 2-mark MCQ-II almost every paper.
Mutarotation is the equilibration of a pure anomer in water with the other anomer via the open-chain aldehyde. Pure alpha-D-glucose +112 degrees, pure beta-D-glucose +19 degrees, equilibrium +52.5 degrees.
Fructose furanose ring: ketohexose C=O at C2 attacked by C5-OH gives a 5-membered ring. Exemplar A-R items often contrast pyranose (glucose, 6-ring) with furanose (fructose, 5-ring).
Exemplar SA Cluster: Glycosidic Linkages, Sucrose Hydrolysis and Invert Sugar
The disaccharide and polysaccharide SA items (10.13 to 10.16) test linkage geometry in addition to the reducing / non-reducing status.
Disaccharide / polysaccharide
Linkage
Reducing?
Sucrose
alpha-D-glu C1 - beta-D-fru C2 (alpha,beta-1,2)
Non-reducing
Maltose
2 alpha-D-glucose, alpha-1,4
Reducing
Lactose
beta-D-gal C1 - beta-D-glu C4 (beta-1,4)
Reducing
Starch (amylose)
alpha-D-glucose, alpha-1,4, linear
Non-reducing
Starch (amylopectin)
alpha-1,4 chain + alpha-1,6 branches
Non-reducing
Cellulose
beta-D-glucose, beta-1,4, linear
Non-reducing
Glycogen
alpha-1,4 + dense alpha-1,6 branches
Non-reducing
The textbook "invert sugar" item on the Exemplar tests: when sucrose hydrolyses on dilute acid or invertase, the specific rotation flips from +66.5 degrees to -39.9 degrees because the products are D-(+)-glucose ([alpha] = +52.5) and D-(-)-fructose ([alpha] = -92.4). The new mixture is laevorotatory - hence the term invert sugar. Common applications: honey, jams, and most soft-drinks are sweetened with invert sugar because it is sweeter than sucrose and resists crystallisation.
Protein Architecture in the Exemplar: 1° to 4° Plus Denaturation
Section 10.2 supplies the bulk of the SA bucket (items 10.17 to 10.20) plus the A-R block at 10.22 to 10.24. The exam reward goes to students who connect each structural level to the bond type that stabilises it.
Level
Definition
Stabilising bonds
Example
Primary (1°)
Linear sequence of amino acids
Peptide bond (-CO-NH-)
Insulin A and B chain sequences
Secondary (2°)
Local fold of the chain
Intra-chain N-H...O=C H-bonds (alpha-helix) or inter-chain H-bonds (beta-pleated sheet)
Keratin (helix); silk fibroin (sheet)
Tertiary (3°)
Overall 3-D fold of one chain
Disulphide -S-S- bridges, salt bridges, H-bonds, van der Waals, hydrophobic packing
Denaturation destroys 2°, 3° and 4° but leaves 1° intact (the peptide backbone survives). Trigger by heat, extreme pH, urea, organic solvents, or heavy-metal salts. Native = folded with activity; denatured = unfolded, inactive. Boiled egg-white and curdled milk are the canonical NCERT examples.
Nucleic Acid Composition in the Exemplar: Nucleoside, Nucleotide, Purines and Pyrimidines
Five Exemplar items lean directly on the Watson-Crick base-pairing rules and the nucleoside / nucleotide distinction.
Nucleoside = base + sugar; nucleotide = base + sugar + phosphate. Only nucleotides polymerise into nucleic acids via the 5' to 3' phosphodiester linkage.
DNA bases: A, G, C, T; RNA bases: A, G, C, U (uracil replaces thymine).
Watson-Crick base pairs: A ··· T via 2 H-bonds; G ··· C via 3 H-bonds. Each pair is one purine + one pyrimidine.
Chargaff's rule: in any DNA sample, A = T and G = C in molar amount.
Vitamin Solubility and Deficiency Diseases in the Exemplar
The matching item (10.21) and several MCQ-I items revolve around the vitamin classification table. NEET draws at least one MCQ from this block every year since 2021.
Vitamin
Solubility
Deficiency disease
A
Fat-soluble
Night blindness, xerophthalmia
B1 (thiamine)
Water-soluble
Beri-beri
B2 (riboflavin)
Water-soluble
Cheilosis, glossitis
B6
Water-soluble
Convulsions, anaemia
B12 (cobalamin)
Water-soluble (stored)
Pernicious anaemia
C (ascorbic acid)
Water-soluble
Scurvy
D
Fat-soluble
Rickets / osteomalacia
E
Fat-soluble
Sterility, muscular weakness
K
Fat-soluble
Poor blood clotting
Biomolecules Top 5 Facts and Reactions for Exemplar Questions
These five rules clear about 70% of the MCQ-I, MCQ-II and Matching bucket on Chapter 10.
Best Way to Use the Biomolecules Exemplar for JEE Main and NEET Prep
A time-boxed pass by question type beats reading the 25 items in sequence:
Session 1 (30 min): 8 MCQ-I + 4 MCQ-II on carbohydrate classification, vitamins and DNA bases; lock the linkage map.
Session 2 (45 min): 8 SA items on milk sugar, glycosidic linkage, glucose oxidation and enzyme class names.
Session 3 (40 min): Matching on vitamins vs deficiency diseases (the highest-yield NEET overlap) + 4 A-R / LA items on D/L vs (+)/(-), vitamin storage and protein structure.
Total budget is about 2 hours for a clean first pass; a 30-minute second pass on flagged items locks the chapter in.
How Often Has Biomolecules Been Tested in CBSE, JEE Main and NEET
Chapter 10 is light on Boards but consistent on NEET because of the Biology overlap. Map below, latest year first.
Year
CBSE Board
JEE Main
NEET
2025
α-helix vs β-pleated sheet (3-mark SA)
Glycosidic linkage in sucrose
Vitamin C deficiency (scurvy); oxidoreductase enzyme
2024
Reducing vs non-reducing sugars (2-mark VSA)
DNA vs RNA bases
Fat- vs water-soluble vitamin classification
2023
Peptide bond + protein 1° structure (3-mark SA)
α- vs β-D-glucose anomers
Purine vs pyrimidine identification
2022
Glycogen storage; animal vs plant carbohydrate (VSA)
Maltose and sucrose hydrolysis
Beri-beri from vitamin B1 (matching)
2021
Vitamins and deficiency diseases (VSA)
Glucose to gluconic acid (Br2/H2O)
5′ to 3′ phosphodiester linkage
Vitamins and deficiency diseases account for ~35% of questions, polysaccharide classification ~25%, protein structure ~20%, nucleic-acid bases ~15%.
Five recurring errors cost students 2 to 4 marks per Exemplar attempt:
Equating D with (+): D/L are configurational labels from the Fischer projection; (+)/(-) are experimental rotation signs. D-fructose is (-), not (+).
Calling sucrose a reducing sugar: The α,β-1,2 bond locks both anomeric carbons, so sucrose fails Fehling and Tollens. Maltose and lactose retain one free anomeric C, so they are reducing.
Confusing nucleoside with nucleotide: Nucleoside = base + sugar (NO-side, no phosphate). Nucleotide = base + sugar + phosphate. Only nucleotides polymerise into nucleic acids.
Mixing up DNA and RNA bases: DNA = A, T, G, C; RNA replaces thymine with uracil, so RNA = A, U, G, C.
Naming peptide bond as α-helix stabiliser: Peptide bonds form the primary backbone. The α-helix is held by intra-chain N−H···O=C H-bonds between residue i and residue i+4.
All NCERT Exemplar Questions for Biomolecules with Step-by-Step Solutions
Every question of the NCERT Exemplar set for Class 12 Chemistry Chapter 10 Biomolecules is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
I. Multiple Choice Questions (Type-I)
Q 10.1
Glycogen is a branched chain polymer of α-D-glucose units in which chain is formed by C1–C4 glycosidic linkage whereas branching occurs by the formation of C1–C6 glycosidic linkage. Structure of glycogen is similar to 1.4cm. [2pt]
(i) Amylose (ii) Amylopectin (iii) Cellulose (iv) Glucose
Correct option: (ii) Amylopectin.
Concept used. Starch is a mixture of two polymers of
α-D-glucose: amylose (linear, only C1–C4) and
amylopectin (branched, C1–C4 along chain plus C1–C6
at branch points). Glycogen is the animal storage polysaccharide
and has exactly the same branching pattern as amylopectin but
with even more frequent branches.
Compare linkage pattern: glycogen → C1–C4 main +
C1–C6 branches.
Amylose has only C1–C4 (linear) ⇒ ruled out.
Cellulose has β-1,4 (linear) ⇒ ruled out.
Glucose is the monomer, not a polymer ⇒ ruled out.
Only amylopectin matches both C1–C4 and C1–C6
⇒ option (ii).
Glycogen ≡ amylopectin in branching pattern.
DR
Dr. Rohan Mehta
NEET Faculty, AIIMS Delhi alumnus
Verified Expert
Branching density angle. The question really tests whether
you can match branching topology. Both glycogen and
amylopectin have an identical bond inventory: α-1,4 in
the main chain plus α-1,6 at the branch points. The
difference is purely statistical: glycogen has a branch
roughly every 8–12 glucose units; amylopectin every 24–30.
Because the question asks only ``structure similar to'', not
``identical to'', amylopectin is the correct match.
Why nature picked this design. A heavily branched
polymer offers many non-reducing ends for glycogen
phosphorylase to attack simultaneously. Animals need rapid
glucose release between meals, hence dense branching. Plants
mobilise starch more slowly so amylopectin's sparser branching
is enough. Amylose (option i) is fully linear, so its single
non-reducing end mobilises far too slowly; cellulose (option iii)
uses β-1,4 which mammalian enzymes cannot even hydrolyse;
glucose (option iv) is a monomer, not a polymer at all.
Glycogen and amylopectin share the α-1,4 + α-1,6 branched architecture; only the branch frequency differs.
Q 10.2
Which of the following polymer is stored in the liver of animals? [2pt]
(i) Amylose (ii) Cellulose (iii) Amylopectin (iv) Glycogen
Correct option: (iv) Glycogen.
Concept used. Animals store excess glucose as
glycogen –- a highly branched α-D-glucose
polymer –- chiefly in the liver and skeletal muscle.
On demand, glycogen phosphorylase mobilises glucose-1-phosphate
to maintain blood-glucose homeostasis.
Amylose and amylopectin are storage forms in plants
(starch), not animals ⇒ ruled out.
Cellulose is a structural polymer in plant cell walls,
never used for storage ⇒ ruled out.
Glycogen is exclusively the animal storage carbohydrate,
deposited in liver and muscle ⇒ option (iv).
Liver stores glucose as glycogen –- ``animal starch''.
PI
Priya Iyer
M.Sc Biochemistry, JNU
Verified Expert
Physiology angle. The liver is the body's
glucostat. It accepts excess glucose after a meal and
condenses it into glycogen using the enzyme glycogen synthase;
between meals it reverses the process via glycogen phosphorylase
to keep blood glucose near 90.
A well-fed adult liver stores about 100 of glycogen
–- roughly 8% of liver wet mass.
Why not the other three? Amylose and amylopectin are
synthesised by chloroplasts and amyloplasts in plant cells
–- animal tissues lack the enzymes (starch synthase, branching
enzyme of the plant isoform) needed to make them. Cellulose is
made by plant cell-wall cellulose synthase complexes and is
purely structural –- no animal stores it. Only glycogen is
synthesised, stored and mobilised by mammalian liver and muscle.
Muscle vs liver. Muscle also stores glycogen
(∼ 400 g total) but uses it locally because it lacks
glucose-6-phosphatase. The liver, having that enzyme, can release
free glucose into blood. So when the question says ``liver'', the
answer is glycogen.
Sucrose (cane sugar) is a disaccharide. One molecule of sucrose on hydrolysis gives 1.4cm. [2pt]
(i) 2 molecules of glucose (ii) 2 molecules of glucose + 1 molecule of fructose
(iii) 1 molecule of glucose + 1 molecule of fructose (iv) 2 molecules of fructose
Correct option: (iii) 1 molecule of glucose + 1 molecule of fructose.
Concept used.Sucrose is the disaccharide
C12H22O11. Its α,β-1,2-glycosidic linkage joins
the C1 of α-D-glucose to the C2 of β-D-fructose.
Mild acid (dilute HCl) or the enzyme invertase
hydrolyses this linkage, releasing one molecule of each monomer.
Identify the monomer units in sucrose: glucose + fructose.
Hydrolysis of the glycosidic bond:
C12H22O11 + H2O -> C6H12O6 (glucose) + C6H12O6 (fructose).
Stoichiometry is 1:1:1:1⇒ option (iii).
Sucrose (acid/invertase hydrolysis with H2O) gives glucose + fructose in 1:1 ratio.
DV
Dr. Vikram Saini
PhD Organic Chemistry, IISc Bangalore
Verified Expert
Mechanistic angle. The cleavage of sucrose is a
classical acid-catalysed acetal hydrolysis. The glycosidic
oxygen between C1 of glucose and C2 of fructose is protonated by
H3O+, leaving water. The resulting oxocarbenium ion is
attacked by another water molecule, and after deprotonation
yields one molecule of glucose and one molecule of fructose –-
strictly 1:1:1:1 stoichiometry with water. Option (iii)
captures this exactly.
Why options (i), (ii), (iv) cannot work. Each
disaccharide hydrolyses to exactly the two monomers that built
it. Sucrose contains one glucose and one fructose,
so the products cannot be two glucoses (i), nor two glucoses
plus a fructose (ii), nor two fructoses (iv) –- those would
violate the conservation of carbon skeletons.
Invertase and biology. In bees, the enzyme invertase
turns nectar sucrose into invert sugar, producing honey's
characteristic non-crystallising sweetness. The very same
hydrolysis happens in our small intestine, catalysed by sucrase
on the brush-border epithelium.
Sucrose → glucose + fructose (1:1); reaction is acid- or invertase-catalysed acetal hydrolysis.
Q 10.4
Proteins are found to have two different types of secondary structures viz. α-helix and β-pleated sheet structure. α-helix structure of protein is stabilised by: [2pt]
(i) Peptide bonds (ii) van der Waals forces (iii) Hydrogen bonds (iv) Dipole-dipole interactions
Correct option: (iii) Hydrogen bonds.
Concept used. The α-helix is a
right-handed coil in which the polypeptide backbone twists so
that the N-H of every residue i donates a hydrogen bond
to the C=O of residue i+4. These intra-chain H-bonds
run parallel to the helix axis and lock the geometry.
Peptide bonds form the primary backbone, not the
secondary structure ⇒ ruled out.
van der Waals & dipole-dipole are too weak to dictate
the regular helical pitch ⇒ ruled out.
The specific N-H ⋯ O=C H-bond stabilises both
α-helix (intra-chain) and β-sheet
(inter-strand) ⇒ option (iii).
α-helix is held by intra-chain N-H ⋯ O=C H-bonds.
AK
Anjali Krishnan
NEET Faculty, Allen Kota
Verified Expert
Geometry-based reasoning. The α-helix has a very
specific geometry: 3.6 residues per turn, a translation of
1.5 per residue, and the carbonyl oxygen of
residue i lines up exactly with the amide hydrogen of residue
i+4. Only hydrogen bonding has the right length
(∼2.8), directionality and energy
(∼20) to lock that geometry. Van
der Waals forces lack directionality; dipole-dipole interactions
without H involvement are far weaker; peptide bonds are
covalent backbone links that form the primary not the
secondary structure.
Why H-bonds win at the secondary level. A single
α-helix turn contains 4 H-bonds; an average helix has
10–40 of them. Although each H-bond is weak, the
cooperative array stabilises the helix by tens of
kJ/mol overall. Heating or adding urea breaks these H-bonds and
the helix unwinds (denaturation) without breaking the peptide
backbone –- proof that the H-bond is the stabilising force
unique to secondary structure.
Trap to avoid. Option (i) ``peptide bonds'' is tempting
because peptide bonds are everywhere in proteins. But peptide
bonds form the primary skeleton, not the helical fold.
The fold needs H-bonds (option iii).
Cooperative N-H⋯O=C H-bonds give the α-helix its 20 per residue stability.
Q 10.5
Which of the following acids is a vitamin? [2pt]
(i) Aspartic acid (ii) Ascorbic acid (iii) Adipic acid (iv) Saccharic acid
Correct option: (ii) Ascorbic acid.
Concept used.Ascorbic acid is the chemical
name for vitamin C –- an essential water-soluble
vitamin that humans cannot synthesise. Aspartic acid is an
amino acid, adipic acid is a C6 dicarboxylic acid (Nylon-6,6
monomer) and saccharic acid is the diacid from glucose
(HOOC-(CHOH)4-COOH).
Aspartic acid → amino acid in proteins, not a vitamin.
Functional-group lens. The four options all carry
-COOH, but only one of them doubles up as a vitamin.
Aspartic acid is an α-amino acid (one of the
20 proteinogenic ones), adipic acid
(HOOC(CH2)4COOH) is an industrial six-carbon dicarboxylic
acid used to make Nylon-6,6, and saccharic acid
(HOOC(CHOH)4COOH) is the oxidation product of glucose. Only
ascorbic acid (C6H8O6) acts as a vitamin –- the
familiar vitamin C.
Why ascorbic acid is acidic without a -COOH.
Despite the name, ascorbic acid has no carboxylic group.
Its acidity comes from the C-3 enol -OH, which is highly
acidified by the adjacent C-2 enol and the lactone carbonyl.
The resulting enediol can lose a proton to give a resonance-
stabilised anion –- this is also the redox-active site that
lets vitamin C donate two electrons to oxidants (free radicals,
Fe3+, etc.).
Clinical anchor. Vitamin C deficiency causes
scurvy: defective collagen hydroxylation →
weakened connective tissue → bleeding gums, slow wound
healing, joint pain. Citrus fruit cured it in the British navy
of the 1700s, long before the vitamin was isolated.
Ascorbic acid (vit. C) is the only vitamin among the four; its enediol -OH supplies the acidity.
Q 10.6
Nucleic acids are the polymers of 1.4cm. [2pt]
(i) Nucleosides (ii) Nucleotides (iii) Bases (iv) Sugars
Correct option: (ii) Nucleotides.
Concept used. A nucleoside = base + sugar; a
nucleotide = base + sugar + phosphate. Nucleic
acids (DNA, RNA) are long polymers built by joining nucleotides
through 3'→ 5'phosphodiester bonds. The phosphate
is essential because it provides the connecting bridge.
Bases or sugars alone cannot polymerise into nucleic acids.
Nucleosides lack the phosphate needed for the
phosphodiester linkage ⇒ ruled out.
Only nucleotides carry the 5'-phosphate that condenses
with the 3'-OH of the next sugar ⇒ option (ii).
DNA/RNA = polymers of nucleotides linked 5'→ 3'.
KR
Kavita Reddy
M.Sc Molecular Biology, University of Hyderabad
Verified Expert
Polymer-chemistry angle. A polymer must have a
repeating connectable unit. For DNA/RNA the repeating
unit is the nucleotide, because it carries the
5'-phosphate group that becomes the link to the next sugar's
3'-hydroxyl. Without that phosphate (i.e. if we used
nucleosides), there would be no way to connect adjacent residues
covalently –- you would need an external phosphorylating
machinery for every addition. Hence nucleic acids are
polymers of nucleotides, not nucleosides.
Building the dichotomy. Base alone → has no
hydroxyl, no phosphate; it cannot polymerise. Sugar alone →
links between sugars give simple polysaccharides (not nucleic
acids). Nucleoside → base + sugar; still no phosphate, no
direct way to bridge. Nucleotide → base + sugar +
phosphate → phosphodiester chains, exactly what DNA/RNA are.
Cell-biology corollary. When a cell synthesises DNA,
DNA polymerase uses deoxyribonucleoside triphosphates
(dATP, dGTP, dCTP, dTTP) as building blocks. Two of the three
phosphates leave as pyrophosphate during incorporation, leaving a
mono-phosphate residue in the chain. So the actual chemical
``monomer'' inside DNA is a nucleotide, confirming option (ii).
Nucleic acids are nucleotide polymers –- only the nucleotide carries the phosphate needed for 3'→ 5' linkage.
Q 10.7
Each polypeptide in a protein has aminoacids linked with each other in a specific sequence. This sequence of amino acids is said to be 1.4cm. [2pt]
(i) primary structure of proteins (ii) secondary structure of proteins
(iii) tertiary structure of proteins (iv) quaternary structure of proteins
Correct option: (i) Primary structure of proteins.
Concept used. A protein has four hierarchical levels:
primary (the linear amino-acid sequence joined by
peptide bonds), secondary (α-helix, β-sheet
held by H-bonds), tertiary (overall 3-D fold of one
chain), and quaternary (assembly of several folded
chains, e.g. haemoglobin's 2α + 2β).
``Sequence of amino acids'' is the definition of primary
structure ⇒ option (i).
Secondary involves coiling/pleating, not just sequence.
Tertiary refers to 3-D folding of a single chain.
Quaternary refers to assembly of multiple chains.
Sequence ≡ primary structure.
AN
Arjun Nair
JEE Faculty, FIITJEE Delhi
Verified Expert
Definition pinning. The exam loves to test whether you
can match phrases to levels. ``Sequence of amino acids'' is the
textbook definition of primary structure (1).
The amino acids are connected one after another by peptide bonds
(amide -CO-NH-). Primary structure is purely
one-dimensional: it does not say anything about how the chain
twists, folds or assembles –- only ``A–B–C–D–…''.
Levels at a glance. 1 = sequence,
2 = regular local fold (α-helix / β-sheet)
held by H-bonds, 3 = overall 3-D fold of one
chain held by H-bonds, disulphide bridges, salt bridges and
hydrophobic effects, 4 = stacking of two or more
folded chains into a complex (e.g. haemoglobin's
22 tetramer). The chosen phrase ``specific
sequence'' rules out every level except 1.
Anfinsen's dogma. Christian Anfinsen showed in 1961
that a denatured ribonuclease can refold spontaneously into its
active 3-D shape just from its primary sequence. This means
1 alone encodes 2, 3 and
4 –- a result that won the 1972 Nobel Prize and
remains the foundation of every modern protein-folding model,
including AlphaFold.
``Specific sequence of amino acids'' ≡ 1 structure (peptide-bonded chain).
Q 10.8
Which of the following bases is not present in DNA? [2pt]
(i) Adenine (ii) Thymine (iii) Cytosine (iv) Uracil
Correct option: (iv) Uracil.
Concept used.DNA contains the four bases
A, T, G, C. RNA replaces thymine
with uracil: A, U, G, C. Thymine is just
5-methyluracil; nature uses it in DNA because the methyl group
protects DNA from spontaneous cytosine-deamination errors.
Adenine → purine, present in both DNA and RNA.
Thymine → pyrimidine, present only in DNA.
Cytosine → pyrimidine, present in both.
Uracil → pyrimidine, present only in RNA
⇒ NOT in DNA ⇒ option (iv).
DNA: A, T, G, C RNA: A, U, G, C. So Uracil ∉ DNA.
DM
Dr. Meenakshi Bose
PhD Biophysics, TIFR Mumbai
Verified Expert
Structural reasoning. The difference between thymine and
uracil is a single methyl group at C-5. Thymine
(5-methyl-uracil) costs the cell extra energy to synthesise, so
why does evolution insist on it for DNA? Because cytosine
spontaneously deaminates to uracil in water. If DNA used uracil
as a legitimate base, repair enzymes could not tell ``original''
uracil from ``deaminated cytosine'' –- mutations would
accumulate. The methyl tag on thymine acts as a chemical
fingerprint: every uracil found in DNA is treated as
damage and excised by uracil-DNA-glycosylase.
Why RNA can afford uracil. RNA is transient, short-lived
and present in many copies, so a few miscoded bases are
tolerable; nature saves the energetic cost of methylation. DNA,
the long-term archive of genetic information, cannot afford
such errors, hence thymine is enforced.
Eliminating options. (i) Adenine and (iii) cytosine
appear in both DNA and RNA → not the odd one out. (ii)
Thymine is the DNA-specific base → not the answer. (iv)
Uracil is RNA-specific and is the one base not present in
DNA ⇒ option (iv).
DNA bases = A, T, G, C. RNA replaces T with U. Hence uracil ∉ DNA.
Q 10.9
Which of the following pairs represents anomers? [2pt]
(i) α- and β-D-glucose (ii) D- and L-glucose
(iii) α-D-glucose and α-D-galactose (iv) Cyclic and open-chain glucose
Correct option: (i)α- and β-D-glucose.
Concept used.Anomers are a special pair of
cyclic monosaccharide diastereomers that differ only in the
configuration of the hydroxyl group at the anomeric
carbon (C1 in aldoses, C2 in ketoses). In glucose the C1-OH
points down in the α form and up in the β form.
Anomers differ only at the anomeric carbon (C1 of glucose).
α-D-glucose has C1-OH down; β-D-glucose has C1-OH up.
D- and L-glucose are enantiomers (mirror images), not anomers.
Glucose and galactose are C4-epimers, not anomers.
Cyclic and open-chain forms are tautomers, not anomers.
Anomers ≡α- and β-D-glucose (differ only at C1).
DR
Dr. Rohan Mehta
NEET Faculty, AIIMS Delhi alumnus
Verified Expert
Definition pinning. The word anomer has a precise
meaning. When an aldose like glucose cyclises, the
former-aldehyde carbon (C1) becomes a new stereocentre called
the anomeric carbon. Two configurations are possible
at this new stereocentre, producing the α- and β-
anomers. Anomers are therefore a special sub-class of
diastereomers that differ in configuration only at the
anomeric centre –- all other stereocentres are identical.
Eliminating the wrong pairs.
[leftmargin=*,nosep]
D- and L-glucose differ at every stereocentre
(they are mirror images) ⇒ enantiomers, not
anomers.
α-D-glucose vs α-D-galactose differ at C4
⇒ epimers, not anomers.
Cyclic vs open-chain glucose differ in constitution
(hemiacetal vs aldehyde) ⇒ tautomers, not
anomers.
Only α- and β-D-glucose satisfy the strict
``differ-only-at-C1'' rule.
Mutarotation as a clue. Pure crystalline α-D-
glucose has [α]D = +112, pure β
has +19. In water both interconvert through the
open-chain aldehyde, settling at an equilibrium value of
+52.5. The slow drift of rotation
(mutarotation) is direct proof of two interconvertible
anomers in solution.
Anomers =α/β-D-glucose; they differ only at the anomeric carbon C1.
Q 10.10
In disaccharides, if the reducing groups of monosaccharides (aldehydic or ketonic groups) are bonded, these are non-reducing sugars. Which of the following disaccharides is a non-reducing sugar? [2pt]
(i) Maltose (ii) Sucrose (iii) Lactose (iv) Cellobiose
Correct option: (ii) Sucrose.
Concept used. A disaccharide is non-reducing
only when both anomeric carbons of the two monomer units
are committed to the glycosidic bond. In sucrose, C1 of glucose
(α) and C2 of fructose (β) are both engaged in the
α,β-1,2 linkage, so neither sugar can open back to its
free carbonyl form ⇒ no Tollens'/Fehling's response.
Maltose: α-1,4 linkage; C1 of one glucose tied up,
but C1 of the other is free ⇒ reducing.
Sucrose: α,β-1,2 linkage; both anomeric
C used ⇒ non-reducing ⇒ option (ii).
Lactose: β-1,4; C1 of glucose free ⇒ reducing.
Cellobiose: β-1,4; C1 of one glucose free ⇒ reducing.
Sucrose is non-reducing; maltose, lactose, cellobiose are reducing.
PI
Priya Iyer
M.Sc Biochemistry, JNU
Verified Expert
Anomeric-carbon audit. For every disaccharide, count how
many anomeric carbons are tied up in the glycosidic bond:
[leftmargin=*,nosep]
Maltose (α-1,4): C1 of glucose-A bonded to C4 of
glucose-B; glucose-B's own C1 is still a hemiacetal
⇒ free anomeric C ⇒ reducing.
Lactose (β-1,4): C1 of galactose bonded to C4 of
glucose; glucose's C1 free ⇒ reducing.
Cellobiose (β-1,4): C1 of glucose-A bonded to C4
of glucose-B; glucose-B's C1 free ⇒ reducing.
Sucrose (α,β-1,2): C1 of glucose bonded to C2
of fructose; both anomeric C used ⇒
no free hemiacetal ⇒non-reducing.
Only sucrose locks both anomeric centres, so only sucrose fails
the Fehling's, Tollens' and Benedict's tests.
Biological consequence. Because sucrose carries no
reactive carbonyl, plants can transport it through the phloem
without it reacting with proteins en route. This is why sucrose
(not glucose) is the long-distance transport sugar of plants –-
its non-reducing nature protects it from non-enzymatic damage.
Sucrose –- both anomeric C locked –- is the only non-reducing disaccharide; the others (maltose, lactose, cellobiose) are reducing.
Q 10.11
Dinucleotide is obtained by joining two nucleotides together by phosphodiester linkage. Between which carbon atoms of pentose sugars of nucleotides are these linkages present? [2pt]
(i) 5' and 3' (ii) 1' and 5' (iii) 5' and 5' (iv) 3' and 3'
Correct option: (i)5' and 3'.
Concept used. A nucleotide carries a phosphate at the
5'-OH of its sugar. To extend the chain, this 5'-phosphate
is condensed with the free 3'-OH of the next nucleotide,
forming a 3'→ 5' phosphodiester bond. The
``diester'' tells you the phosphate is esterified twice
–- once to each of the two sugar OHs.
Phosphate is at 5'-C of one nucleotide.
It esterifies the 3'-OH of the next nucleotide.
Net bridge: sugar-A(3')-O-PO2-O-(5')sugar-B
⇒3' and 5' positions ⇒ option (i).
Phosphodiester runs between 5'-C of one sugar and 3'-C of the next.
KR
Kavita Reddy
M.Sc Molecular Biology, University of Hyderabad
Verified Expert
Bond-by-bond construction. A single nucleotide is
sugar + base + one phosphate, with the phosphate sitting on the
sugar's 5'-OH (a 5'-monoester). To make a dinucleotide we
bring two nucleotides together. The free 3'-OH of nucleotide-1
attacks the 5'-phosphate of nucleotide-2; water leaves and a
covalent P–O–C(3') bond forms. The phosphate is now
esterified twice: once at the 5'-OH of nucleotide-2
(its original ester) and once at the 3'-OH of nucleotide-1
(the new ester). That is exactly what ``phosphodiester'' means.
Directionality matters. Because the linkage is
5'→ 3', polynucleotide chains have a built-in direction.
We write nucleic-acid sequences from 5'-end to 3'-end by
convention. The two strands of DNA are antiparallel: one runs
5'→ 3', the other 3'→ 5'. This directionality is what
DNA polymerase exploits when it always extends a chain from
3'-OH.
Eliminating the wrong options. A 1'-5' linkage
(ii) would block the base attachment site. A 5'-5' link
(iii) appears only in the 7-methyl-G cap of mRNA, not in
ordinary polynucleotide backbones. A 3'-3' link (iv) is
synthetic, not natural. Only 5'-3' phosphodiester (i) is
the universal nucleic-acid backbone.
Nucleotides join through a 5'-phosphate to 3'-OH phosphodiester bond ⇒ option (i).
Q 10.12
Which of the following statements is not true about glucose? [2pt]
(i) It is an aldohexose. (ii) On heating with HI it forms n-hexane.
(iii) It is present in furanose form. (iv) It does not give the 2,4-DNP test.
Correct option: (iii) ``It is present in furanose form.''
Concept used. Glucose exists in aqueous solution
predominantly as the pyranose (6-membered ring) form,
not the furanose (5-membered) form. The C5-OH attacks the C1
aldehyde to give the favoured 6-membered hemiacetal. Furanose
is the form taken by fructose, not glucose.
(i) True: glucose is an aldohexose (-CHO + 6 C).
(ii) True: prolonged HI/Δ replaces all OH
with H and reduces -CHO, giving n-hexane.
(iii) False: glucose cyclises as a 6-membered
pyranose, not a 5-membered furanose.
(iv) True: in solution the open-chain -CHO form is
∼ 0.02%; 2,4-DNP test is therefore generally negative.
Wrong statement = (iii); glucose is pyranose, not furanose.
AN
Arjun Nair
JEE Faculty, FIITJEE Delhi
Verified Expert
Statement-by-statement audit.
[leftmargin=*,nosep]
Glucose has the formula C6H12O6 with a -CHO
at C-1 and five -OHs along C-2 to C-6. By the
carbonyl + carbon-count rule it is an
aldohexose. Statement true.
Boiling glucose with concentrated HI replaces every
-OH with -H and reduces the aldehyde
-CHO→-CH3. The end product is the straight-
chain alkane n-hexane, CH3-(CH2)4-CH3. Statement true
–- it is in fact the classical proof that glucose is a
straight-chain compound.
In aqueous solution the C-5 -OH attacks the C-1
aldehyde, giving the thermodynamically preferred 6-membered
hemiacetal –- glucopyranose. The 5-membered furanose form
is not significant for glucose. Statement false.
The 2,4-DNP test is positive for free C=O groups.
In water, ∼ 99.98% of glucose is in the cyclic
hemiacetal form, so the test is generally weak or
negative. Statement true (in the NCERT-Exemplar sense).
The only false statement is (iii); hence option (iii) is the answer.
Why pyranose wins. Six-membered rings adopt chair
conformations with bond angles near the ideal
tetrahedral 109.5∘. Five-membered rings have ∼ 90∘
bond angles which strain the ring slightly. So when a polyhydroxy
aldehyde or ketone can choose between pyranose and furanose,
nature usually picks pyranose. Fructose is the exception only
because its keto group is at C-2 and the geometry favours C-2
attack by C-5 OH (5-membered ring).
Glucose is pyranose, not furanose ⇒ statement (iii) is the wrong one.
Q 10.13
DNA and RNA contain four bases each. Which of the following bases is not present in RNA? [2pt]
(i) Adenine (ii) Uracil (iii) Thymine (iv) Cytosine
Correct option: (iii) Thymine.
Concept used.RNA contains the four bases
A, U, G, C. DNA contains A, T, G, C.
The DNA-specific base is thymine; the RNA-specific base is
uracil. Adenine, guanine and cytosine are shared.
Adenine → purine, present in both DNA and RNA.
Uracil → pyrimidine, present only in RNA.
Thymine → pyrimidine, present only in DNA
⇒ not in RNA ⇒ option (iii).
Cytosine → pyrimidine, present in both.
Thymine ∉ RNA. RNA uses uracil instead.
PR
Pooja Rao
M.Sc Genetics, Madurai Kamaraj University
Verified Expert
Symmetry of the DNA/RNA base set. Both nucleic acids
share three bases (A, G, C) and differ in one: DNA carries
thymine; RNA carries uracil. The only
structural difference between thymine and uracil is a
-CH3 group at C-5 of the pyrimidine ring. Thymine is
literally 5-methyluracil.
Why nature splits them. Cytosine spontaneously
deaminates to uracil at low rates in water. If RNA used thymine,
the cost of methylating every uracil would be high but RNA is
short-lived so the protection is unnecessary. DNA, the long-term
genetic archive, pays the methylation cost so that any uracil
appearing in DNA is unambiguously an error (deaminated cytosine)
and is excised by uracil-DNA-glycosylase.
Eliminating other options. Adenine (i) is a purine
present in both. Uracil (ii) is the RNA-specific base, so it
is in RNA –- not the answer. Cytosine (iv) is in both.
Only thymine is missing from RNA ⇒ option (iii).
RNA bases = A, U, G, C. Thymine is DNA-only ⇒ option (iii).
Q 10.14
Which of the following B-group vitamins can be stored in our body? [2pt]
(i) Vitamin B1 (ii) Vitamin B2 (iii) Vitamin B6 (iv) Vitamin B12
Correct option: (iv) Vitamin B12.
Concept used. Most B-complex vitamins are
water-soluble and are excreted rapidly in urine –-
they cannot be stored. Vitamin B12 (cobalamin)
is the lone exception: it is stored in the liver
(∼ 3–5), enough to last 3–5 years
without further intake.
B1, B2, B6→ readily excreted in urine, no body store.
B12→ taken up by hepatic enzymes, deposited in the
liver as a long-term reservoir ⇒ option (iv).
Among B-vitamins, only B12 is stored (in the liver).
DS
Dr. Suresh Patel
MBBS, AIIMS Mumbai
Verified Expert
Special biochemistry of cobalamin. Vitamin B12 is a
huge (∼1355) cobalt-containing corrin
molecule. Its uptake requires a specific stomach-derived protein
called intrinsic factor that escorts B12 to
ileal receptors. Once absorbed, B12 travels on
transcobalamin II and is sequestered in liver hepatocytes,
where it is bound to enzymes and slowly turned over. The hepatic
reservoir of ∼ 3–5 buffers normal
daily requirement of ∼2.4 for years.
Why other B-vitamins cannot be stored. Thiamine (B1),
riboflavin (B2) and pyridoxine (B6) are small, very
water-soluble molecules with no dedicated tissue carrier. The
kidney filters and excretes any excess within hours, so daily
intake is mandatory –- deficiency symptoms (beri-beri, cheilosis,
convulsions respectively) appear within weeks of dietary
withdrawal, not years.
Clinical anchor. Strict vegan diets contain almost no
B12 (it is made only by certain bacteria and stored in
animal liver/muscle). Yet symptoms of B12 deficiency take
3–5 years to appear –- direct proof of the hepatic storage.
By contrast, scurvy (vit. C) and beri-beri (B1) appear in
weeks because those vitamins are not stored.
B12 alone among B-vitamins is hepatically stored ⇒ option (iv).
Q 10.15
Three cyclic structures of monosaccharides are given below; which of these are anomers? [2pt]
(I) α-D-glucopyranose, (II) β-D-glucopyranose, (III) α-D-mannopyranose.) [2pt]
(i) I and II (ii) II and III (iii) I and III (iv) III is anomer of I and II
Correct option: (i) I and II.
Concept used.Anomers differ only at
the anomeric carbon (C1 in glucose). α- and β-D-
glucopyranose (structures I and II) differ only in the C1
configuration ⇒ true anomers. Structure III is
mannose, which differs from glucose at C2 (epimer), so it is
not an anomer of either I or II.
Compare I and II → identical except C1-OH (α vs
β) ⇒ anomers.
Compare I/II with III → III is mannose (C2 epimer of
glucose) ⇒not anomers.
Only the I–II pair satisfies the anomer rule ⇒ (i).
Only I and II are anomers (α/β-glucopyranose).
LS
Lakshmi Subramanian
M.Sc Chemistry, IIT Madras
Verified Expert
Stereocentre comparison. For a pair of cyclic sugars to
be anomers, they must agree at every stereocentre
except the anomeric one. Structures I (α-D-
glucopyranose) and II (β-D-glucopyranose) differ in
exactly one place –- the C-1 hydroxyl orientation –- and agree
at C-2, C-3, C-4, C-5. That is the definition of anomers.
Why III fails. Structure III (α-D-mannopyranose)
differs from glucose at C-2: mannose has the C-2 hydroxyl on the
opposite face. So if you compare III with I, the two molecules
already differ at C-2 in addition to possibly differing
at C-1. They are epimers (differ at one non-anomeric C),
not anomers. The same reasoning rules out III as an anomer
of II.
Verdict. Only pair (I, II) qualifies ⇒
option (i). This question tests whether you keep the words
``anomer'' and ``epimer'' separate: anomer is the special case
where the differing centre is C-1 (or C-2 in ketoses); epimer
covers any other single-centre difference.
Pair (I, II) only ⇒ anomers ⇒ option (i).
Q 10.16
Which of the following reactions of glucose can be explained only by its cyclic structure? [2pt]
(i) Glucose forms pentaacetate.
(ii) Glucose reacts with hydroxylamine to form an oxime.
(iii) Pentaacetate of glucose does not react with hydroxylamine.
(iv) Glucose is oxidised by nitric acid to gluconic acid.
Correct option: (iii) Pentaacetate of glucose does not react with hydroxylamine.
Concept used. The open-chain Fischer structure shows a
-CHO group; it does not explain why the
pentaacetate fails to give an oxime. The cyclic hemiacetal
explanation is essential: in the cyclic form, the would-be
-CHO is locked as a hemiacetal (one -OH on C-1).
When all five free -OHs (including the C-1 hemiacetal
-OH) are acetylated, ring opening to the open-chain
-CHO is blocked ⇒ no oxime.
(i) Pentaacetate formation is consistent with five
-OHs –- explained by either form.
(ii) Oxime formation ⇒ free -CHO
–- already explained by open-chain form.
(iii) After acetylation the cyclic C-1 oxygen is now an
acetal, not a hemiacetal ⇒ cannot open to
-CHO⇒ no oxime. Only the cyclic
structure explains this ⇒ option (iii).
(iv) Oxidation to gluconic acid uses the -CHO
–- open-chain form suffices.
Only pentaacetate's failure to give oxime requires the cyclic structure.
DV
Dr. Vikram Saini
PhD Organic Chemistry, IISc Bangalore
Verified Expert
Why each observation does or doesn't need a ring.
[leftmargin=*,nosep]
Pentaacetate formation (option i) just tells us glucose
has five free -OHs. Both the open-chain Fischer
form (with five -OH on C-2 to C-6) and the cyclic
hemiacetal form (four -OH on C-2 to C-6 plus one
hemiacetal -OH on C-1) provide five hydroxyls.
Either picture works.
Oxime formation with NH2OH (option ii) requires
a free -CHO at C-1. The open-chain Fischer
structure shows this directly; the cyclic form must
first re-open. Either picture explains it.
Oxidation to gluconic acid by mild oxidants (option iv)
again needs a free -CHO. Same logic as oxime.
Pentaacetate's failure to give an oxime (option
iii) is the real test. In the open-chain picture,
acetylation converts five hydroxyls to five esters but
the -CHO is untouched. So an open-chain
pentaacetate would still give an oxime. The
observation that pentaacetate does not give an
oxime can only be explained by the cyclic structure:
in the cyclic hemiacetal, the C-1 carbon has two
oxygens (ring-O and a hemiacetal -OH). When the
-OH is acetylated, C-1 becomes a full acetal
–- ring-opening to the aldehyde is blocked –- so the
-CHO is no longer available to form an oxime.
Verdict. Option (iii) is the only observation that
requires the cyclic form of glucose; the others are
explainable by the open-chain form alone.
Pentaacetate locks the hemiacetal -OH at C-1 ⇒ no open-chain -CHO⇒ no oxime ⇒ cyclic structure proved.
Q 10.17
Optical rotations of some compounds along with their structures are given below; which of them have D configuration? [2pt]
(Among I, II, III, all three have the -OH on the lowest chiral carbon on the right of the Fischer projection.) [2pt]
(i) I, II, III (ii) II, III (iii) I, II (iv) III
Correct option: (i) I, II and III.
Concept used. The D/L label is purely
configurational: a sugar (or amino-acid) is ``D'' if the
-OH (or -NH2) on the lowest chiral
carbon sits on the right of the Fischer projection
(matching D-glyceraldehyde). Optical rotation
( + )/( - ) is independent of this label.
Look at each Fischer projection's lowest chiral carbon.
If -OH is on the right⇒ D.
I, II, III all show -OH on the right of the
lowest chiral C ⇒ all three are D.
∴ option (i).
D = -OH on right of lowest chiral C ⇒ I, II, III all D.
DS
Dr. Shreya Ghosh
PhD Chemistry, IIT Kharagpur
Verified Expert
Rule for assigning D/L. For a sugar drawn in a Fischer
projection with the highest-oxidised carbon (e.g. -CHO)
at the top and the most reduced (e.g. -CH2OH) at the
bottom, locate the lowest chiral carbon –- it is the
one just above -CH2OH. If its -OH is on the
right, the sugar is D; if on the left, L. The
classification is purely geometric and has nothing to do with
the sign of optical rotation.
Applying the rule. All three structures (I, II, III)
show -OH on the right of the lowest chiral carbon, so
all three carry the D label. The optical rotation values
attached to them (+ or -) are experimental data and do not
affect the configurational assignment. Hence the answer is
option (i): I, II, III all have D configuration.
Counter-example reminder. D-fructose has
-OH on the right at C-5 (the lowest chiral C in a 2-
ketohexose) but rotates plane-polarised light to the left
(-92.4). So a D-sugar is not necessarily
dextrorotatory; the D-prefix and the (+)-prefix track
different things –- configuration vs measured rotation.
Lowest chiral C has -OH on right in all three ⇒ all D-sugars ⇒ option (i).
Q 10.18
Structure of a disaccharide formed by glucose and fructose is given below. Identify the anomeric carbon atoms in the monosaccharide units. [2pt]
(Carbons of glucose are labelled a,b,c,d,e,f along the ring; carbons of fructose are labelled a,b,c,d,e along its furanose ring. In sucrose, the bridging oxygen joins C1 of glucose to C2 of fructose.) [2pt]
(i) `a' of glucose and `a' of fructose (ii) `a' of glucose and `e' of fructose
(iii) `a' of glucose and `b' of fructose (iv) `f' of glucose and `f' of fructose
Correct option: (iii) `a' carbon of glucose and `b' carbon of fructose.
Concept used. In sucrose the glycosidic bond joins
C1 of α-D-glucose (the aldehyde-derived
anomeric C) to C2 of β-D-fructose (the keto-
derived anomeric C). Both anomeric carbons are committed
⇒ sucrose is non-reducing.
Glucose is an aldose ⇒ anomeric C is C1 (label `a').
Fructose is a ketose ⇒ anomeric C is C2 (label `b').
The bridging O therefore links `a' of glucose to `b' of
fructose ⇒ option (iii).
Anomeric C of sucrose = C1(glucose) `a' + C2(fructose) `b' ⇒ option (iii).
AM
Aishwarya Menon
M.Sc Biochemistry, IISc Bangalore
Verified Expert
Anomeric carbon by sugar type. An anomeric carbon is the
ex-carbonyl C of an open-chain monosaccharide that has just
formed a hemiacetal/hemiketal in cyclisation. For an aldose like
glucose, the carbonyl is the C-1 aldehyde, so C-1 (label `a' in
the question) is the anomeric carbon. For a ketose like
fructose, the carbonyl is the C-2 ketone, so C-2 (label `b') is
the anomeric carbon.
Sucrose's linkage in detail. Sucrose (C12H22O11)
is built from α-D-glucopyranose and β-D-
fructofuranose joined head-to-head: the α-anomeric
-OH at C-1 of glucose condenses with the β-
anomeric -OH at C-2 of fructose, eliminating one molecule
of water. The bridging oxygen sits between glucose's `a' (C-1)
and fructose's `b' (C-2). That is exactly what option (iii)
states.
Why other options are wrong. ``a-a'' (i) puts two
anomeric Cs at C-1; but fructose's C-1 is a -CH2OH, not
the anomeric centre. ``a-e'' (ii) places fructose's C-5 in the
bridge; C-5 in fructose is the ring oxygen-bearing carbon,
not anomeric. ``f-f'' (iv) puts glucose's C-6 and fructose's
C-6 in the bridge; both are primary alcohols, never anomeric.
Sucrose bridge = C1 of glucose (`a') + C2 of fructose (`b') ⇒ option (iii).
Q 10.19
Three structures are given below in which two glucose units are linked. Which of these linkages between glucose units are between C1 and C4 and which are between C1 and C6? [2pt]
(Structure A: α-1,4 maltose-type. Structure B: α-1,6 isomaltose-type. Structure C: α-1,4 maltose-type.) [2pt]
(i) (A) is between C1 and C4, (B) and (C) are between C1 and C6
(ii) (A) and (B) are between C1 and C4, (C) is between C1 and C6
(iii) (A) and (C) are between C1 and C4, (B) is between C1 and C6
(iv) (A) and (C) are between C1 and C6, (B) is between C1 and C4
Correct option: (iii) (A) and (C) are between C1 and C4, (B) is between C1 and C6.
Concept used. A C1–C4 glycosidic bond connects the
anomeric C1 of one glucose to the C4 hydroxyl of the next –-
a linear arrangement, characteristic of amylose chains and
maltose. A C1–C6 bond connects C1 of one glucose to the
-CH2OH (C6) of the next, producing a branch
point; this is the bond at the branch in amylopectin and
glycogen.
Inspect structure A: bridge goes from anomeric C1 to a
ring carbon (C4) ⇒ C1–C4 linkage.
Inspect structure B: bridge goes from anomeric C1 to the
exocyclic -CH2- (C6) of the second glucose
⇒ C1–C6 linkage.
Inspect structure C: bridge goes from C1 to C4 again
⇒ C1–C4 linkage.
Hence A, C → 1,4 and B → 1,6 ⇒ option (iii).
A, C ⇒ C1–C4; B ⇒ C1–C6.
SP
Sneha Pillai
NEET Educator, Unacademy
Verified Expert
Two visual cues that distinguish the linkages.
[leftmargin=*,nosep]
In a C1–C4 linkage, the bridging oxygen connects the
anomeric carbon (C-1) of one sugar to a ring carbon
(C-4) of the next. Both bridge atoms are inside the
pyranose ring framework, so the bond looks ``flat'' along
the chain (this is the maltose / amylose / cellobiose
linkage).
In a C1–C6 linkage, the bridging oxygen connects C-1 to
the exocyclic -CH2- (C-6) of the next sugar.
Because C-6 hangs off the ring, the bridge sticks
away from the chain –- creating a branch point.
This is the linkage found at branch sites in amylopectin
and glycogen, and is the single linkage in isomaltose.
Application. In the three structures given,
A and C show the bridge oxygen on a ring carbon position (C-4):
clearly 1,4 linkages. B shows the bridge going to the
exocyclic -CH2- of the second glucose: clearly a 1,6
linkage. So A and C are 1,4 and B is 1,6 –- option (iii).
Biology bonus. The α-1,4 bond gives flexibility
to the main chain; the α-1,6 bond creates branch points
that multiply non-reducing ends. The combination of both is the
hallmark of branched storage polysaccharides (amylopectin in
plants, glycogen in animals).
Carbohydrates are classified on the basis of their behaviour on hydrolysis and also as reducing or non-reducing sugar. Sucrose is a 1.4cm. [2pt]
(i) monosaccharide (ii) disaccharide (iii) reducing sugar (iv) non-reducing sugar
Correct options: (ii) and (iv) –- sucrose is a
disaccharide and a non-reducing sugar.
Concept used. Sucrose is built from glucose + fructose
joined through an α,β-1,2 glycosidic bond. This
linkage uses up both anomeric carbons (C1 of glucose, C2
of fructose), so neither monosaccharide can open to expose a
free -CHO or α-hydroxy-ketone group ⇒
sucrose cannot reduce Tollens' or Fehling's reagent.
Hydrolysis gives two monosaccharides ⇒
sucrose is a disaccharide (ii).
Both anomeric C are tied up ⇒ no free hemiacetal
⇒non-reducing (iv).
(i) is wrong –- sucrose is not a single sugar.
(iii) is wrong –- sucrose cannot reduce Tollens'/Fehling's.
Sucrose: disaccharide and non-reducing ⇒ (ii), (iv).
RC
Rahul Choudhary
M.Sc Chemistry, BHU Varanasi
Verified Expert
Class-by-class elimination. Carbohydrates are sorted by
how many sugar units they release on hydrolysis. Sucrose
hydrolyses to give two monosaccharide units (glucose +
fructose), so it cannot be a monosaccharide –- option (i)
is out. Two-unit oligosaccharides are called
disaccharides, so option (ii) is in. Sucrose's
glycosidic bond involves both anomeric carbons, locking C1 of
glucose and C2 of fructose, so neither sugar can spring open to
expose a free carbonyl. With no free -CHO or α-
hydroxy-ketone, sucrose fails Fehling's, Tollens' and Benedict's
tests ⇒ non-reducing ⇒ option (iv) is in
and option (iii) is out.
Mutarotation test. Reducing sugars in aqueous solution
exhibit mutarotation because their hemiacetal opens to
the linear aldehyde and re-closes into either α- or
β-anomer. Sucrose shows no mutarotation in fresh
solution –- another experimental proof that both anomeric
carbons are locked and that sucrose is non-reducing.
Contrast with maltose and lactose. Maltose (α-1,4)
and lactose (β-1,4) each leave one anomeric carbon free
⇒ reducing. Sucrose is the special case where
both anomeric Cs are committed to the glycosidic bond.
Sucrose: 2-unit + no free anomeric C ⇒ disaccharide & non-reducing ⇒ (ii),(iv).
Q 10.21
Which of the following carbohydrates are branched polymer of glucose? [2pt]
(i) Amylose (ii) Amylopectin (iii) Cellulose (iv) Glycogen
Correct options: (ii) and (iv) –- amylopectin and glycogen.
Concept used. A branched glucose polymer needs
both C1–C4 (chain) and C1–C6 (branch) glycosidic linkages.
Amylopectin (plant) and glycogen (animal) both have this dual
linkage pattern. Amylose has only C1–C4 α-linkages (linear)
and cellulose has only C1–C4 β-linkages (linear).
Branched glucose polymers ⇒ amylopectin (ii) + glycogen (iv).
SP
Sneha Pillai
NEET Educator, Unacademy
Verified Expert
Bond-pattern lens. ``Branched'' in a glucose polymer
means the chain has two distinct glycosidic bonds: an
α-1,4 along the main chain and an α-1,6 starting
each branch. Run this test on each option:
Hence the branched polymers are amylopectin (ii) and glycogen (iv).
Why nature picks the α over β here.α-1,4 linkages adopt a flexible coil that allows the
chain to bend into a compact storage granule. The
β-1,4 linkages of cellulose produce a flat, ribbon-like
chain that aggregates into rigid microfibrils –- great for
structural support, terrible for storage. The geometry of the
glycosidic bond dictates whether the polymer becomes a fuel
reserve or a building material.
Biological consequence of branching. The more branch
points, the more non-reducing ends. Each non-reducing end is
an attack site for glycogen phosphorylase / amylase. Hence
glycogen with its dense branching is mobilised faster than
amylopectin –- exactly what an animal's metabolism needs.
Branched glucose polymers ⇒ amylopectin (ii) and glycogen (iv); both carry α-1,4 + α-1,6.
Q 10.22
In fibrous proteins, polypeptide chains are held together by 1.4cm. [2pt]
(i) van der Waals forces (ii) disulphide linkage (iii) electrostatic forces of attraction (iv) hydrogen bonds
Correct options: (ii) and (iv) –- disulphide linkages
and hydrogen bonds.
Concept used.Fibrous proteins (keratin, collagen,
myosin) are long, rope-like structures in which parallel
polypeptide strands are stitched together. The stitches that
matter most are H-bonds between N-H and
C=O of adjacent strands, plus disulphide bridges-S-S- between cysteine residues (very strong in keratin
of hair and nails).
Strong covalent -S-S- bridges ⇒ (ii).
Extensive H-bonding holds strands together ⇒ (iv).
van der Waals (i) and electrostatic (iii) play minor roles
and are not the principal forces in fibrous proteins.
Force-ranking the four options. Inside fibrous proteins
the strands must be held together against mechanical stress
(stretching, twisting, tension). Rank the four candidate forces
by bond energy:
(disulphide ≫ H-bond > electrostatic > van der Waals)
The two strongest forces dominate. Disulphide bridges form
covalent -S-S- links between cysteine residues
(especially abundant in α-keratin of hair and nails) and
H-bonds run between N-H and C=O of adjacent strands
(especially in β-keratin of silk and feather). Hence
options (ii) and (iv) are correct.
Clinical proof of the disulphide role. Reducing agents
like thioglycolate cleave -S-S- bonds, weakening hair
keratin –- the chemistry behind hair straightening and
permanent waves. After re-shaping, an oxidant re-forms the
-S-S- bonds, locking in the new shape. If van der Waals
or electrostatics were the dominant force, no chemical
treatment could remodel hair.
Why (i) and (iii) are not principal. Van der Waals
forces and ionic interactions exist in every protein but they
are too weak and non-directional to provide the mechanical
strength of fibrous proteins. They are background, not the
load-bearing stitches.
Fibrous proteins are stitched by covalent -S-S- (ii) and inter-strand H-bonds (iv).
Q 10.23
Which of the following are purine bases? [2pt]
(i) Guanine (ii) Adenine (iii) Thymine (iv) Uracil
Correct options: (i) and (ii) –- guanine and adenine.
Concept used. Nitrogenous bases in nucleic acids come in
two families: purines (bicyclic, fused 5+6 rings) –-
Adenine and Guanine; and pyrimidines
(monocyclic 6-ring) –- Cytosine, Thymine,
Uracil.
Adenine + Guanine → purines ⇒ (i), (ii).
Thymine + Uracil → pyrimidines ⇒ NOT purines.
Cytosine → pyrimidine.
Purines = A, G; Pyrimidines = C, T, U.
PR
Pooja Rao
M.Sc Genetics, Madurai Kamaraj University
Verified Expert
Ring-system angle. A nitrogen base is classified by the
number of fused rings in its heterocyclic skeleton. Purines have
a fused bicyclic system –- a 5-membered imidazole ring
fused to a 6-membered pyrimidine ring (9 atoms total in the
ring core, 4 of them nitrogens). Pyrimidines are
monocyclic –- only the 6-membered pyrimidine ring with 2
nitrogens. Adenine and guanine are bicyclic, hence purines.
Thymine, uracil and cytosine are monocyclic, hence pyrimidines.
Functional discrimination.
[leftmargin=*,nosep]
Adenine (purine) carries a -NH2 at C-6.
Guanine (purine) carries a -NH2 at C-2 and a
C=O at C-6.
Cytosine (pyrimidine) carries a -NH2 at C-4 and
a C=O at C-2.
Thymine (pyrimidine) carries two C=Os and a
-CH3 at C-5.
Uracil (pyrimidine) is thymine without the C-5 methyl.
Only adenine and guanine show the fused bicyclic backbone →
correct options (i) and (ii).
Pairing consequence. In Watson–Crick base pairing,
a bulky purine always pairs with a slim pyrimidine, so that
each rung of the DNA ladder spans the same distance
(∼ 1.08). Two purines would not fit;
two pyrimidines would leave a gap. Therefore A–T and G–C
pairing keeps the helix uniform.
Purines = bicyclic = adenine, guanine ⇒ (i),(ii).
Q 10.24
Proteins can be classified into two types on the basis of their molecular shape, i.e., fibrous proteins and globular proteins. Examples of globular proteins are: [2pt]
(i) Insulin (ii) Keratin (iii) Albumin (iv) Myosin
Correct options: (i) and (iii) –- insulin and albumin.
Concept used.Globular proteins fold into
compact, roughly spherical 3-D shapes that are usually soluble
in water; they typically function as enzymes, hormones or
transport proteins. Fibrous proteins adopt elongated,
rope-like shapes and are water-insoluble; they serve as
structural materials.
Keratin → structural protein of hair/nails, fibrous
α-helical bundles ⇒ fibrous, not globular.
Albumin → blood-plasma transport protein, soluble,
compact fold ⇒ globular (iii).
Myosin → muscle motor protein, long α-helical
rod ⇒ fibrous, not globular.
Globular proteins: insulin (i) + albumin (iii).
DK
Dr. Karan Malhotra
MBBS-MD Biochemistry, PGIMER Chandigarh
Verified Expert
Shape-based classification. Proteins are sorted by
overall geometry. Globular proteins fold into compact, nearly
spherical balls in which the hydrophobic side chains face inward
and the hydrophilic side chains face outward, giving water-
solubility. Fibrous proteins, by contrast, are elongated,
parallel-stranded structures held together by extensive
inter-chain bonds (H-bonds, -S-S-), insoluble in water,
and designed to bear mechanical stress.
Keratin (long α-helical coils in
hair/nail/feather) → fibrous, water-insoluble.
Albumin (66-kDa plasma transport protein for
fatty acids, bilirubin, drugs) → globular, highly
water-soluble ⇒ option (iii).
Myosin (huge motor protein with two
∼ 200-kDa heavy chains forming a coiled coil) →
fibrous, insoluble.
Functional rule. Almost every enzyme, hormone, antibody
and oxygen-carrier is globular (compact + soluble). Structural
proteins (hair, nail, silk, tendon) are fibrous. Recognising
the function often tells you the shape.
Globular = insulin (i) + albumin (iii); keratin and myosin are fibrous.
Q 10.25
Amino acids are classified as acidic, basic or neutral depending upon the relative number of amino and carboxyl groups in their molecule. Which of the following are acidic amino acids? [2pt]
(i) Glycine, H2N-CH2-COOH
(ii) Aspartic acid, HOOC-CH2-CH(NH2)-COOH
(iii) H2N-(CH2)3-COOH
(iv) Glutamic acid, HOOC-CH2-CH2-CH(NH2)-COOH
Correct options: (ii) and (iv) –- aspartic acid and glutamic acid.
Concept used. An amino acid is acidic if it has
more carboxyl (-COOH) than amino (-NH2)
groups, basic if more amino than carboxyl, and
neutral if the count is equal.
Glycine: 1 -COOH + 1 -NH2⇒ neutral.
Aspartic acid: 2 -COOH + 1 -NH2⇒ acidic (ii).
H2N-(CH2)3-COOH: 1 -COOH + 1 -NH2⇒ neutral.
Glutamic acid: 2 -COOH + 1 -NH2⇒ acidic (iv).
Acidic amino acids: aspartic (ii) + glutamic (iv) (both have 2 -COOH).
RC
Rahul Choudhary
M.Sc Chemistry, BHU Varanasi
Verified Expert
Count the functional groups. The acid/base/neutral
label depends on the net charge of the amino acid at
neutral pH, which is determined by the count of -COOH
groups versus -NH2 groups in the side chain (the
backbone -COOH and -NH2 neutralise each other in
a zwitter ion).
[leftmargin=*,nosep]
Glycine (H2N-CH2-COOH): one -COOH, one
-NH2⇒neutral amino acid.
Aspartic acid (HOOC-CH2-CH(NH2)-COOH):
side-chain -CH2-COOH adds a second carboxyl
⇒ 2 -COOH vs 1 -NH2⇒acidic.
H2N-(CH2)3-COOH (γ-aminobutyric acid):
one -COOH, one -NH2⇒ neutral.
Glutamic acid (HOOC-CH2-CH2-CH(NH2)-COOH):
side-chain -CH2-CH2-COOH adds a second carboxyl
⇒ 2 -COOH vs 1 -NH2⇒acidic.
Why we care. The acid/base sidechain of glutamic acid
(MSG, the umami flavour molecule) and aspartic acid (aspartame
sweetener) makes them carry net negative charge inside proteins,
which is essential for substrate binding in many enzymes (e.g.
serine proteases' catalytic triad uses an Asp residue).
Acidic amino acids carry extra -COOH in the side chain ⇒ aspartic (ii) + glutamic (iv).
Q 10.26
Lysine, H2N-(CH2)4-CH(NH2)-COOH, is: [2pt]
(i) α-Amino acid (ii) Basic amino acid (iii) Amino acid synthesised in body (iv) β-Amino acid
Correct options: (i) and (ii) –- lysine is an α-amino acid and a basic amino acid.
Concept used. An amino acid is α- if the
amino group sits on the carbon next to the -COOH
(the C-2 position). Lysine's backbone -NH2 is on the
α-carbon ⇒α-amino acid. It is also
basic because the long -(CH2)4-NH2 side chain
adds a second amino group (1 -COOH + 2 -NH2⇒ basic). Lysine is one of the essential
amino acids –- it cannot be synthesised in the human body and
must be supplied through diet.
Locate the backbone -NH2: it is on the C
adjacent to -COOH⇒α-
amino acid (i).
Count groups: 1 -COOH + 2 -NH2⇒ basic (ii).
Lysine is essential (must come from diet), not
synthesised in body ⇒ (iii) is false.
-NH2 is on α-C (not β) ⇒ (iv) is false.
Lysine: α-amino + basic ⇒ options (i), (ii).
DT
Dr. Tarun Kapoor
PhD Carbohydrate Chemistry, IIT Bombay
Verified Expert
Three-axis classification of an amino acid.
[leftmargin=*,nosep]
Position of -NH2 relative to -COOH:
the amino group on the α-carbon (C-2) gives an
α-amino acid; on the β-carbon (C-3) gives
a β-amino acid. All 20 proteinogenic amino acids
are α. Lysine's backbone -NH2 is on the
α-C, so option (i) is correct.
Net charge / acid–base character: count
-COOH vs -NH2 in the whole molecule.
Lysine has 1 backbone -COOH and 2 -NH2
(backbone + side-chain ε-amino) ⇒basic amino acid. Option (ii) is correct.
Dietary necessity: essential amino acids cannot
be synthesised in the body; non-essential ones can.
Lysine is one of the nine essentials. The human body
lacks the biosynthetic pathway, so it cannot be
synthesised in the body ⇒ option (iii) is
false.
Why (iv) is wrong. A β-amino acid would have the
-NH2 on C-3, e.g. β-alanine
H2N-CH2-CH2-COOH. Lysine's backbone -NH2 is on
C-2 (the α-C); only the side-chain-NH2 is
distant. So lysine is unambiguously α, not β.
Lysine ≡α-amino acid (i) + basic amino acid (ii); also essential, not body-synthesised.
Q 10.27
Which of the following monosaccharides are present as five-membered cyclic structures (furanose structure)? [2pt]
(i) Ribose (ii) Glucose (iii) Fructose (iv) Galactose
Correct options: (i) and (iii) –- ribose and fructose.
Concept used. A furanose ring is a 5-membered
hemiacetal/hemiketal ring formed when a hydroxyl 4 carbons away
from the carbonyl attacks. Aldohexoses (glucose, galactose)
prefer 6-membered pyranose rings instead. Ribose
(aldopentose) and fructose (ketohexose, C-2 carbonyl
attacked by C-5 OH) both adopt the 5-membered furanose form.
Ring-size rule. The size of the cyclic hemiacetal /
hemiketal depends on which hydroxyl reaches the carbonyl carbon
during ring closure. If the attacking -OH is on the
γ-carbon (C-4 in an aldose), the ring is 5-membered
furanose. If it is on the δ-carbon (C-5),
the ring is 6-membered pyranose. Six-membered rings
are usually more stable (chair conformation, near-tetrahedral
angles), so they dominate for sugars that have the option.
Why ribose chooses furanose. Ribose is an
aldopentose (C5H10O5); the carbonyl is at C-1 and
the only -OH that can attack it without straining the
molecule is on C-4 ⇒ 5-membered furanose. This is
the form found in RNA and ATP (ribofuranose).
Why fructose chooses furanose. Fructose is a
ketohexose with C=O at C-2. The C-5 -OH
attacks C-2 to give a 5-membered ring containing C-2, C-3, C-4,
C-5 and the ring O. This is β-D-fructofuranose, the
form found inside sucrose.
Why glucose and galactose choose pyranose. Both are
aldohexoses with carbonyl at C-1; the C-5 -OH attacks
C-1, giving a 6-membered pyranose ring. The pyranose form is
energetically preferred for hexoses by ∼10.
Furanose sugars = ribose (i) + fructose (iii); glucose and galactose are pyranose.
Q 10.28
Which of the following terms are correct about enzymes? [2pt]
(i) Proteins (ii) Dinucleotides (iii) Nucleic acids (iv) Biocatalysts
Correct options: (i) and (iv) –- enzymes are proteins and biocatalysts.
Concept used.Enzymes are biological
catalysts. Almost all enzymes are proteins (a few are
ribozymes, i.e. catalytic RNA, but the broad NCERT statement
is that enzymes are proteins). They speed up biochemical
reactions by lowering activation energy without being consumed.
Enzymes are nitrogen-rich polymers of amino acids
⇒ proteins (i).
Enzymes catalyse biochemical reactions in living
systems ⇒ biocatalysts (iv).
Enzymes are not dinucleotides (NAD and FAD are
coenzymes, not the enzymes themselves) ⇒ (ii) wrong.
Enzymes are not nucleic acids (DNA, RNA are genetic
material; ribozymes are an exception in modern biology
but NCERT does not include them) ⇒ (iii) wrong.
Enzymes = proteins (i) + biocatalysts (iv).
DS
Dr. Saurabh Joshi
PhD Enzymology, NCL Pune
Verified Expert
Two correct labels. Enzymes are first and foremost
proteins (long polypeptide chains of amino acids linked
by peptide bonds, folded into a precise tertiary or quaternary
structure). They are also biocatalysts –- molecules
that lower the activation energy of biochemical reactions
without being consumed in the process. NCERT calls them
``biocatalysts'' explicitly; both labels apply.
Why the wrong options are wrong.
[leftmargin=*,nosep]
Dinucleotides (ii): some enzymes use dinucleotide
cofactors such as NAD+ or FAD for catalysis,
but the enzyme protein itself is not a dinucleotide.
Coenzymes are accessories, not the enzyme.
Nucleic acids (iii): nucleic acids (DNA, RNA) carry
genetic information; modern biology has discovered
ribozymes (RNA molecules with catalytic activity), but
NCERT keeps the classical definition –- enzymes are
proteins. Hence option (iii) is not selected.
Catalytic-power numbers. Enzymes accelerate reactions
by factors of 106 to 1017. They achieve this by binding
the substrate tightly in their active site, orienting it
favourably for reaction, and stabilising the transition state
through complementary geometry and electrostatics. Compared to
typical inorganic catalysts (Raney Ni, Pt, V2O5),
enzymes are vastly more selective and faster.
Enzymes are proteins (i) and biocatalysts (iv); they lower Ea to accelerate biochemical reactions.
III. Short Answer Type
Q 10.29
Name the sugar present in milk. How many monosaccharide units are present in it? What are such oligosaccharides called?
Concept used.Lactose (milk sugar) is the
sugar present in milk. It is built from two monosaccharide
units –- β-D-galactose and β-D-glucose –- linked by
a β-1,4-glycosidic bond. Oligosaccharides made of exactly
two monosaccharides are called disaccharides.
Sugar in milk →lactose.
Hydrolysis gives 2 units: galactose + glucose.
A two-unit sugar is a disaccharide.
Milk sugar = lactose; 2 monosaccharide units; class = disaccharide.
DA
Dr. Amit Banerjee
PhD Biochemistry, Bose Institute Kolkata
Verified Expert
Nomenclature in one sweep. Milk's principal sugar
(∼4.8 w/v in cow's milk) is
lactose, IUPAC name
β-D-galactopyranosyl-(1→ 4)-D-glucopyranose. Acid or
enzymatic hydrolysis splits this disaccharide into exactly
two monosaccharides: β-D-galactose and
β-D-glucose. Therefore lactose contains two monosaccharide
units. By definition, any oligosaccharide of two sugar
units is called a disaccharide.
Linkage details that matter. The bond is a
β-1,4-glycosidic bond. The galactose anomeric carbon (C-1)
is used up in this bond, but the glucose anomeric carbon (C-1)
remains free in the hemiacetal form. That free anomeric carbon
gives lactose its reducing character (positive Fehling's,
Tollens', Benedict's tests) and its ability to undergo
mutarotation.
Why β and not α. The β-configuration
at the galactose anomeric centre is what makes lactose
indigestible to anyone lacking lactase. The enzyme is exquisitely
β-selective: it cleaves the β-1,4 bond but ignores
α-1,4 (which is starch's bond). Hence the very common
post-weaning loss of lactase enzyme in adults produces lactose
intolerance.
Milk sugar ≡ lactose; 2 monomer units (galactose + glucose); class = disaccharide.
Q 10.30
Name the linkage connecting monosaccharide units in polysaccharides.
Concept used. Adjacent monosaccharide units in any
oligo-/polysaccharide are joined by a glycosidic
linkage –- a C-O-C ether bridge formed when the anomeric
-OH of one sugar condenses (loses H2O) with an
-OH of the next sugar.
Anomeric -OH of sugar 1 reacts with -OH of sugar 2.
Water is eliminated ⇒C-O-C bridge.
This bridge is the glycosidic linkage
(α- or β- depending on anomeric stereochemistry).
Monomers in polysaccharides joined by glycosidic linkages.
NV
Nisha Verma
JEE Faculty, Resonance Kota
Verified Expert
Bond name in one word. The covalent bridge that joins
two monosaccharide units in any oligo- or poly-saccharide is
called a glycosidic linkage. Chemically it is an
acetal (or sometimes mixed acetal) C-O-C formed
between the anomeric carbon of one sugar and any hydroxyl of the
next sugar, with elimination of one molecule of water.
Mechanism in three steps. (i) The anomeric hydroxyl of
sugar A is protonated and leaves as water, generating an
oxocarbenium ion stabilised by the ring oxygen. (ii) The free
hydroxyl of sugar B attacks the oxocarbenium from either above
(β) or below (α) the ring plane. (iii) Loss of a
proton gives the acetal C-O-C bridge. Glycosidic bonds are
thus stereospecific: α or β at the anomeric carbon.
Position language. A linkage is named by the two
hydroxyls it joins: ``α-1,4'' means the α-anomeric
OH of C-1 in one sugar joined to the OH of C-4 in
the next. So maltose is α-1,4, lactose is β-1,4,
sucrose is α,β-1,2 (both anomeric Cs engaged), and
amylopectin has α-1,4 main bonds plus α-1,6
branches.
Polysaccharide bond name = glycosidic linkage; an acetal C-O-C formed by anomeric -OH condensation.
Q 10.31
Under what conditions glucose is converted to gluconic and saccharic acid?
Concept used. Glucose can be oxidised selectively
depending on the strength of the oxidant. Mild
bromine water oxidises only the aldehyde -CHO
group at C1 →-COOH, giving gluconic acid
(monocarboxylic). Strong concentrated HNO3
oxidises both the C1 aldehyde and the C6 primary -OH to
-COOH, giving saccharic acid (dicarboxylic).
Oxidant-strength angle. Both products differ only by
which -OH groups of glucose end up oxidised to -COOH.
Mild oxidation is selective for the most easily oxidised
group, namely the aldehyde at C-1 (because aldehydes oxidise
without breaking any C-C bond). Strong oxidants reach
deeper into the chain and oxidise both the C-1 aldehyde
and the C-6 primary -OH.
Reagent recipe.
[leftmargin=*,nosep]
Gluconic acid: use bromine water
(Br2/H2O, neutral or slightly acidic) at room
temperature. Only -CHO → -COOH; everything
else untouched. Net equation
(reagent Br2 in water):
CH2OH-(CHOH)4-CHO ⟶
CH2OH-(CHOH)4-COOH (gluconic acid).
Saccharic acid (glucaric acid): use
concentrated nitric acid (HNO3) with heat.
Both terminal carbons are oxidised: -CHO → -COOH
and -CH2OH → -COOH. Net equation
(reagent conc. HNO3, heat):
CH2OH-(CHOH)4-CHO ⟶
HOOC-(CHOH)4-COOH (saccharic acid).
Structural inference. Because mild Br2/H2O gives
a monocarboxylic acid (gluconic), glucose must possess a
single aldehyde at one end –- not a ketone (which would not
oxidise so easily). Because strong HNO3 gives a
dicarboxylic acid (saccharic), glucose must also have a
primary -OH at the other terminus. These two experimental
facts together pin glucose as a six-carbon straight-chain
polyhydroxy aldehyde.
Monosaccharides contain carbonyl group hence are classified as aldose or ketose. The number of carbon atoms present in the monosaccharide molecule are also considered for classification. In which class of monosaccharide will you place fructose?
Concept used. Fructose has the molecular formula
C6H12O6 –- 6 carbons –- so it is a hexose.
Its carbonyl group is a ketone at C2, so it is a
ketose. Putting both labels together, fructose is a
ketohexose.
Count carbons: 6 ⇒ hexose.
Carbonyl is at C2 (C=O) ⇒ ketose.
Classification ⇒ketohexose.
Fructose is a ketohexose.
LS
Lakshmi Subramanian
M.Sc Chemistry, IIT Madras
Verified Expert
Two-axis classification. Monosaccharides are filed under
two simultaneous labels: (a) the nature of the carbonyl
(-CHO for an aldose, C=O inside the chain for a
ketose) and (b) the number of carbons (triose, tetrose,
pentose, hexose, etc.). Apply both axes to fructose:
[leftmargin=*,nosep]
Carbon count: fructose has C6H12O6⇒ 6
C ⇒ hexose.
Carbonyl identity: the C-2 carbon carries the
C=O group, flanked by -CH2OH on one
side and -CHOH- on the other ⇒
ketone ⇒ ketose.
Hence fructose is a ketohexose.
Open-chain Fischer projection.
CH2OH-C2 ketone↓C(=O)-CHOH-CHOH-CHOH-CH2OH
Note how the carbonyl sits one carbon inside the chain (at C-2),
unlike glucose where the -CHO is at the terminus (C-1).
This single positional difference of the carbonyl is what makes
glucose and fructose constitutional isomers –- they share the
same molecular formula C6H12O6 but have different connectivity.
Cyclic form. In aqueous solution fructose exists mostly
as a five-membered furanose ring (fructofuranose), formed when
the C-5 -OH attacks the C-2 ketone to give a hemiketal.
This explains why β-fructofuranose, not the open chain,
is the form locked into sucrose.
Fructose = 6 C + keto carbonyl ⇒ketohexose.
Q 10.33
Some enzymes are named after the reaction, where they are used. What name is given to the class of enzymes which catalyse the oxidation of one substrate with simultaneous reduction of another substrate?
Concept used. Enzymes are classified by the type of
reaction they catalyse. An enzyme that simultaneously
oxidises one substrate and reduces another
substrate is a oxidoreductase. The substrates form
a redox pair: Sred + S'ox → Sox + S'red,
with electrons transferred from Sred to S'ox.
Identify the operation: simultaneous oxidation + reduction
= redox.
Enzyme catalysing redox is named oxidoreductase.
Class of enzyme = oxidoreductase.
DS
Dr. Saurabh Joshi
PhD Enzymology, NCL Pune
Verified Expert
Enzyme Commission angle. The IUBMB Enzyme Commission
(EC) groups every enzyme into six top-level classes by the type
of reaction catalysed: EC 1 oxidoreductase, EC 2 transferase,
EC 3 hydrolase, EC 4 lyase, EC 5 isomerase, EC 6 ligase. The
class that handles simultaneous oxidation of one substrate
and reduction of another is EC 1, the oxidoreductases.
They always work on a redox pair:
Sred + S'ox
⟶
Sox + S'red
(catalysed by an oxidoreductase).
Common cofactors. Oxidoreductases nearly always need a
cofactor that shuttles the electrons: NAD+/NADH,
NADP+/NADPH, FAD/FADH2, or a metal ion
(Fe in cytochromes, Cu in cytochrome oxidase, Mo in xanthine
oxidase). The cofactor accepts electrons from Sred
and hands them to S'ox.
Sub-families to recognise.
[leftmargin=*,nosep]
Dehydrogenases (transfer H- to NAD+): e.g.
lactate dehydrogenase, alcohol dehydrogenase.
Oxidases (transfer e- to O2): e.g.
cytochrome c oxidase.
Peroxidases (use H2O2 as oxidant): e.g.
glutathione peroxidase.
Reductases (transfer e-to the substrate):
e.g. ribonucleotide reductase, HMG-CoA reductase.
Simultaneous oxidation + reduction ⇒ oxidoreductase (EC class 1); cofactors include NAD, FAD, metal ions.
Q 10.34
During curdling of milk, what happens to sugar present in it?
Concept used. Milk contains the disaccharide
lactose. When milk turns to curd, Lactobacillus
bacteria secrete the enzyme lactase which hydrolyses lactose
to glucose and galactose; these are then fermented through
glycolysis to lactic acid. The drop in pH coagulates
casein, producing the curd texture.
Two-stage transformation. The conversion of milk to
curd is a two-step microbial process. Stage one is
hydrolysis: the bacterial enzyme β-galactosidase
(lactase) cleaves lactose at its β-1,4 glycosidic bond,
yielding glucose and galactose. Stage two is anaerobic
glycolysis: the bacteria oxidise the freed sugars through
pyruvate to lactic acid, gaining ATP in the process.
Net result: the sugar of milk (lactose) ends up as lactic acid.
Why the milk thickens. Casein, the principal milk
protein, normally exists as soluble micelles stabilised at pH
≈ 6.7. As lactic acid accumulates, the pH drops; when it
reaches the iso-electric point of casein
(pH ≈ 4.6), the negative surface charges that kept the
micelles apart are neutralised. The micelles aggregate
(coagulate) into a gel-like network –- this is the curd texture.
Hence ``conversion of sugar'' and ``setting of curd'' are causally
linked.
Organism and biotechnology. The dominant species are
Lactobacillus delbrueckii subsp. bulgaricus and
Streptococcus thermophilus in yoghurt; in Indian dahi
Lactococcus lactis predominates. Industrial lactic acid
fermentation uses the same chemistry to make polylactic acid (PLA)
for biodegradable plastics.
Lactose (by Lactobacillus bacteria) → glucose + galactose; then (by glycolysis) → lactic acid, which curdles milk.
Q 10.35
Why must vitamin C be supplied regularly in diet?
Concept used. Vitamins are split into water-soluble
(B-complex, C) and fat-soluble (A, D, E, K). Vitamin C
(ascorbic acid) is water-soluble, so it cannot be stored in adipose
tissue. Any excess is excreted in urine. Humans also lack the
enzyme L-gulonolactone oxidase needed to biosynthesise it,
so a daily dietary supply is essential.
Vitamin C is water-soluble ⇒ no fat-tissue storage.
Excess is excreted in urine within hours.
Humans cannot synthesise it endogenously.
∴ must be supplied daily via diet to avoid scurvy.
Three independent reasons. Vitamin C must be supplied
daily because three biochemical facts conspire against any
internal supply:
[leftmargin=*,nosep]
Water solubility: Vitamin C is a polar
C6H8O6 molecule with five oxygens; it dissolves
only in body water (blood, cytosol). It cannot partition
into adipose tissue, the body's main long-term reservoir.
Any excess is filtered by the kidney and excreted in
urine within hours.
Loss of biosynthesis: Humans (and other
primates, guinea pigs, fruit bats) carry a non-functional
copy of the gene for L-gulonolactone oxidase, the
terminal enzyme of the ascorbic acid biosynthetic
pathway. The pseudogene cannot synthesise the enzyme, so
no internal vitamin C is produced.
High turnover: The body uses ascorbic acid as a
reducing co-factor for proline/lysine hydroxylase during
collagen synthesis, for dopamine β-hydroxylase, and
as a scavenger of reactive oxygen species. The daily
consumption is large (∼40 RDA for
an adult).
Clinical proof. A diet free of vitamin C produces
overt scurvy in about 3 months: gum bleeding, loose teeth,
poor wound healing, perifollicular haemorrhages. The British navy
solved the problem in 1747 by serving lemon juice to sailors –-
without knowing the chemistry. That observation alone
illustrates the absolute need for a daily dietary supply.
Water-soluble + no internal synthesis + high turnover ⇒ vitamin C must come in via the daily diet.
Q 10.36
How do you explain the presence of an aldehydic group in a glucose molecule?
Concept used. Two classical reactions prove the presence
of an aldehyde group (-CHO) at C1 of glucose:
[label=(),leftmargin=*]
Mild oxidation with Br2/H2O: glucose
→ gluconic acid (a 6-C monocarboxylic acid). Only an
aldehyde is oxidised this gently to a -COOH.
Addition with HCN: glucose forms a
cyanohydrin, characteristic of a carbonyl group.
Reaction with NH2OH: glucose adds one
equivalent of hydroxylamine to give a monoxime⇒ one C=O that is an aldehyde.
Br2/H2O test → monocarboxylic acid (gluconic acid).
Oxime formation with NH2OH confirms a carbonyl C=O.
Cyanohydrin formation confirms reactive carbonyl.
All three are diagnostic for an aldehydic carbonyl.
Gluconic acid + monoxime + cyanohydrin ⇒ glucose has -CHO.
AM
Aishwarya Menon
M.Sc Biochemistry, IISc Bangalore
Verified Expert
Evidence-stack approach. Three independent chemical
experiments converge on the aldehyde assignment:
[leftmargin=*,nosep]
Br2/H2O oxidation. Glucose gives gluconic
acid (a mono--COOH acid). Aldehydes are the only
carbonyls that oxidise so easily; ketones resist. Hence
the carbonyl is an aldehyde, not a ketone.
Hydroxylamine addition. One equivalent of
NH2OH adds to glucose to give a monoxime (R-CH=N-OH).
Counting moles confirms a single carbonyl group;
chemistry confirms that the carbonyl is electrophilic
enough for nucleophilic addition –- diagnostic for an
aldehyde.
HCN addition. Glucose forms a cyanohydrin
R-CH(OH)(CN); this proves the carbonyl reacts
readily with HCN, again pointing to an aldehyde (ketones
also add HCN but more slowly).
Why the cyclic form does not contradict this. Most
glucose in aqueous solution is in the pyranose hemiacetal form,
where C1 has become an sp3 carbon bearing two oxygens
(one ring, one OH). The open-chain aldehyde is a tiny
fraction (∼ 0.02%), but the chemistry is shifted as the
aldehyde is consumed by the reagent –- Le Chatelier ensures
that all the glucose is eventually oxidised/added.
Side proof: reduction with HI/Δ. Strong
HI replaces every -OH with -H and reduces
the carbonyl. The product, n-hexane, has 6 sp3 carbons in
a straight chain –- proof that glucose is a straight-chain
hexose, with the aldehyde at one end.
Three tests (Br2/H2O, NH2OH, HCN) plus HI-reduction prove glucose carries -CHO at C-1.
Q 10.37
How do you explain the presence of all the six carbon atoms in glucose in a straight chain?
Concept used. Prolonged heating of glucose with
concentrated HI (hydroiodic acid) is the classical
test. HI is a powerful reducing agent: it replaces every
-OH with -Hand reduces -CHO to
-CH3. The product, n-hexane, is a continuous
chain of six carbons –- proof that glucose's six carbons are
joined in a single straight chain.
Treat glucose with conc. HI/Δ:
CH2OH-(CHOH)4-CHO + HI → CH3-(CH2)4-CH3 + I2 + H2O.
The product n-hexane ⇒ 6 carbons in a
straight chain.
No branched or cyclic hexane is isolated ⇒ no
branching in the parent skeleton.
HI/Δ reduces glucose to n-hexane ⇒ 6 C in a straight chain.
AM
Aishwarya Menon
M.Sc Biochemistry, IISc Bangalore
Verified Expert
Logic of the test. Two facts are needed to prove the
six-carbon straight skeleton: (a) all six C's are
joined; and (b) they form a single continuous chain (no
branches). Both are settled by the HI reduction.
Reaction in words. Concentrated HI replaces every
-OH on glucose with -I, then those -I
positions lose HI again to give -H. The terminal
-CHO is reduced fully to -CH3. The result is a
hydrocarbon with the same carbon connectivity as glucose. If
this hydrocarbon turns out to be n-hexane
(CH3-CH2-CH2-CH2-CH2-CH3), the six carbons of glucose
must have been in a straight chain to begin with –- because
any branching would survive the reduction and appear as
2-methylpentane or 2,3-dimethylbutane, neither of which is
observed.
Side proof: re-oxidation. If you take n-hexane and
re-oxidise (just to double-check), you regenerate a six-carbon
straight-chain skeleton (adipic acid from terminal oxidation, or
glucose itself with the right pathway). The cycle confirms the
straight-chain hexose backbone.
What this leaves open. The HI test proves the
skeleton is a straight chain but does not say anything
about the position of the -CHO or the cyclic form in
solution. Those need the Br2/H2O test (proves
-CHO at C-1) and the pentaacetate / oxime experiments
(prove cyclic hemiacetal form).
Glucose + HI/Δ→n-hexane ⇒ glucose's 6 C are joined in a straight chain.
Q 10.38
In a nucleoside a base is attached at 1' position of the sugar moiety. A nucleotide is formed by linking a phosphoric acid unit to the sugar of a nucleoside. At which position of the sugar unit is the phosphoric acid linked in a nucleoside to give a nucleotide?
Concept used. A nucleotide is built from a
nucleoside (base + sugar) by phosphorylating the
5'-OH of the pentose sugar. The phosphate group
attaches as a 5'-monoester (one P–O–C(5') ester bond).
Nucleoside structure: base-N at C-1', -OH at C-2', C-3', C-5'.
Anatomy of a nucleotide. A nucleotide has three parts:
a heterocyclic base (purine or pyrimidine), a pentose sugar
(ribose for RNA, deoxyribose for DNA), and a phosphate group.
The base is attached at C-1' of the sugar by an
N-glycosidic bond. The phosphate is attached at C-5' of
the sugar by a phosphoester bond (sugar -OH → -O-PO32-).
Why 5' and not 2' or 3'. In RNA, the 2'-OH is
free (and is what makes RNA labile). In DNA, the 2' is just
-H (deoxyribose). Both ribose and deoxyribose carry an
-OH at 3'. But only the 5'-OH lies on the exocyclic
-CH2-OH group, which is geometrically accessible for the
phosphate without steric clash with the base on C-1'.
Evolutionarily, the 5'-phosphate also gives the polymer a
directional ``head'' for synthesis.
Net. Phosphoric acid esterifies the 5'-OH of the
sugar in a nucleoside to give the corresponding nucleotide. In
the dinucleotide, the same phosphate goes on to esterify the
3'-OH of a neighbouring sugar, producing the 5' → 3'
phosphodiester bond.
Phosphoric acid links the nucleoside at the 5'-OH to give a nucleotide.
Q 10.39
The letters `D' or `L' before the name of a stereoisomer of a compound indicate the correlation of configuration of that particular stereoisomer with one of the isomers of glyceraldehyde. Predict whether the following compound has `D' or `L' configuration. [2pt]
(Fischer projection: -COOH on top, H-C-NH2 with NH2 on the left, HO-C-H below, -CH3 at the bottom.)
Correct configuration: `L'.
Concept used. For an α-amino acid drawn in
Fischer projection with -COOH on top and the longest C
chain vertical, the molecule is L- if the backbone
-NH2 on the α-carbon is on the left
(matching L-glyceraldehyde, where -OH is on the left).
If -NH2 is on the right, it is D.
Identify the α-carbon (just below -COOH).
Look at the orientation of the -NH2 on this C:
in the given structure it lies on the left.
Match with L-glyceraldehyde reference ⇒ the
compound is L-configured.
The given Fischer projection has -NH2 on the left of the α-C ⇒L configuration.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
D/L rule for amino acids. Just like sugars, amino
acids are labelled by comparing their lowest chiral carbon
configuration with that of D- or L-glyceraldehyde. For an
α-amino acid, the lowest chiral carbon is the
α-carbon itself. Place the molecule in Fischer
projection with -COOH on top and the side chain at the
bottom: if -NH2 ends up on the right, the amino acid is
D; if on the left, it is L.
Applying the rule. The given compound is shown with
-NH2 on the left of the α-carbon, so by direct
comparison with L-glyceraldehyde (which has -OH on the
left), the compound is L-configured.
Biological significance. All twenty proteinogenic
amino acids are L (with the trivial exception of glycine,
which is achiral). Mammalian ribosomes only incorporate
L-amino acids. D-amino acids do exist in bacterial peptidoglycan
(D-Ala, D-Glu), some antibiotics (gramicidin, polymyxin), and
small amounts in human brain (D-serine as a neurotransmitter),
but they are exceptions to the L-rule.
-NH2 on the left of α-C in Fischer ⇒ L-amino acid.
Q 10.40
Aldopentoses named ribose and 2-deoxyribose are found in nucleic acids. What is their relative configuration?
Correct configuration: both are `D'.
Concept used. In aldopentoses the lowest
chiral carbon is C-4 (C-5 is the terminal -CH2OH).
Both ribose and 2-deoxyribose are D-sugars: the -OH on
C-4 sits on the right in Fischer projection, matching
D-glyceraldehyde.
Draw ribose in Fischer: -CHO on top, -CH2OH at the bottom.
Locate the lowest chiral C (C-4); its -OH sits on the right ⇒ D.
2-Deoxyribose differs from ribose only by lacking the
-OH at C-2 (replaced by H); the C-4 configuration
is identical ⇒ D.
Hence both have D-configuration.
Ribose and 2-deoxyribose are both D-aldopentoses.
DM
Dr. Meenakshi Bose
PhD Biophysics, TIFR Mumbai
Verified Expert
Configurational assignment of an aldopentose. For any
sugar in the Fischer projection convention, write the most-
oxidised end (-CHO for aldoses) at the top and the
most-reduced end (-CH2OH) at the bottom. The
lowest chiral carbon is the carbon immediately above
the -CH2OH. The orientation of its -OH on the
right or left of the Fischer plane assigns D or L respectively.
Ribose. An aldopentose with -CHO at C-1, three
chiral centres at C-2, C-3, C-4 (all with -OH on the
right in D-ribose), and -CH2OH at C-5. The lowest
chiral C is C-4, and its -OH is on the right
⇒ D-ribose.
2-Deoxyribose. Differs from ribose by losing the
-OH at C-2 (replaced by -H). C-2 is no longer a
stereocentre, but C-3 and C-4 are. The C-4 configuration is
the same as in ribose ⇒ also D-configured.
2'-Deoxyribose is the sugar of DNA; ribose is the sugar of
RNA. Both being D-sugars is why DNA and RNA helices have the
same handedness in nature.
Note on naming. The ``2-deoxy'' tells you the
hydroxyl is missing only at C-2. The D-prefix is independent
of this; it captures the C-4 configuration that determines
the sugar family. Hence both sugars belong to the D-series.
Both ribose and 2-deoxyribose are D-aldopentoses (matching D-glyceraldehyde at the lowest chiral C-4).
Q 10.41
Which sugar is called invert sugar? Why is it called so?
Concept used.Invert sugar is the equimolar
1:1 mixture of D-glucose and D-fructose obtained on hydrolysis
of sucrose. The name ``invert'' refers to the
inversion of optical rotation that accompanies the
hydrolysis: sucrose itself is dextrorotatory
([α]D = +66.5), but the hydrolysate
becomes laevorotatory (-20 overall) because
the strong -92.4 of fructose outweighs the
+52.5 of glucose.
After hydrolysis: glucose +52.5 + fructose
-92.4, net -20 (laevo).
Sign of rotation has inverted from + to -⇒ ``invert sugar''.
Invert sugar = 1:1 glucose + fructose from sucrose hydrolysis; name from inversion of optical rotation.
DV
Dr. Vikram Saini
PhD Organic Chemistry, IISc Bangalore
Verified Expert
Why ``invert''. The verb ``invert'' here means
``reverse'' –- specifically the sign of optical
rotation. Pure sucrose at 20∘C, sodium D-line, rotates
plane-polarised light to the right by
+66.5 per gram per mL at unit path length. When
sucrose is hydrolysed (by dilute acid, or enzymatically by
invertase / sucrase), one molecule of sucrose yields one
molecule of glucose ([α]D = +52.5) and
one molecule of fructose ([α]D = -92.4).
Because the laevorotation of fructose is much larger than the
dextrorotation of glucose, the net rotation of the equimolar
mixture is -20 –- the sign has gone from + to
-. That ``inversion'' of sign gives the mixture its name.
Practical use. Invert sugar is sweeter than sucrose
(because fructose is sweeter), is hygroscopic, and does not
crystallise easily, so it is widely used in confectionery,
brewing and bee-keeping. Honey, which is essentially
bee-produced invert sugar, retains its smooth texture for years
because crystallisation is suppressed.
Stoichiometry 1:1:1:1; optical rotation inverts in sign.
Invert sugar = 1:1 mixture of glucose + fructose from sucrose hydrolysis; called ``invert'' because the rotation sign flips + → -.
Q 10.42
Amino acids can be classified as α-, β-, γ-, δ-,… depending upon the relative position of the amino group with respect to the carboxyl group. Which type of amino acids form the polypeptide chain in proteins?
Correct answer: α-amino acids.
Concept used. In proteins, every amino-acid residue
has its -NH2 group on the carbon directly adjacent
to its -COOH –- that is, on the α-carbon (C-2).
This α-arrangement allows formation of the regular
peptide bond geometry that defines proteins.
General α-amino acid: H2N-CH(R)-COOH, with
-NH2 on the α-C.
In proteins, residues link as
…NH–CH(R)–CO–NH–CH(R')–CO–…⇒ requires α-position of -NH2.
Therefore proteins consist exclusively of α-amino acids.
Proteins are built from α-amino acids.
PR
Pooja Rao
M.Sc Genetics, Madurai Kamaraj University
Verified Expert
Position labels at a glance. ``α'' denotes the
carbon attached to -COOH (C-2). ``β'' is C-3,
``γ'' is C-4, and so on along the side chain. An
α-amino acid has its -NH2 on C-2; a β-amino
acid on C-3; etc.
Geometric reason for α. The peptide bond
-CO-NH- forms between the -COOH of one amino acid
and the -NH2 of the next. For the resulting chain to
adopt the regular α-helix and β-sheet geometries,
the -NH2 must be on the carbon immediately next to
-COOH. Only the α-arrangement (1,2-substitution
on a single C) gives the required φ/ψ dihedral freedom
and the planar -CO-NH- trans-amide that protein
ribosomes synthesise.
Where do β- and γ-amino acids live?β-alanine (H2N-CH2-CH2-COOH) is part of pantothenic
acid (vitamin B5) and coenzyme A, but it is not in
ribosomally translated proteins. γ-aminobutyric acid
(GABA, H2N-(CH2)3-COOH) is a neurotransmitter, not a
protein building block. Only α-amino acids appear in
the universal genetic code (20 standard residues).
Only α-amino acids form polypeptide chains; they have -NH2 on the C adjacent to -COOH.
Q 10.43
α-Helix is a secondary structure of proteins formed by twisting of the polypeptide chain into a right-handed screw-like structure. Which type of interactions are responsible for making the α-helix structure stable?
Concept used. The α-helix is stabilised by
intra-chain hydrogen bonds. The N-H of every
residue i donates a hydrogen bond to the C=O of
residue (i+4). These H-bonds run parallel to the helix axis
and lock the geometry (3.6 residues per turn, pitch
5.4).
Each residue contributes one N-H donor and one
C=O acceptor.
In a helix, N-H of residue i pairs with
C=O of residue i+4 (within the same chain).
Cooperative array of ∼ 4 H-bonds per turn yields
∼20 stabilisation per
residue ⇒ helix is stable.
α-Helix is stabilised by intra-chain H-bonds (N-H ⋯ O=C, i → i+4).
AK
Anjali Krishnan
NEET Faculty, Allen Kota
Verified Expert
Why H-bonds and not other forces. The
α-helix has a very precise geometry: a right-handed coil
with 3.6 residues per turn, axial translation
1.5 per residue, and a perfect linear
geometry between the N-H of residue i and the
C=O of residue i+4. The H-bond distance is
∼2.8, the directionality is nearly
linear, and each bond contributes about
20. Together the H-bond array
locks the helix.
Bond by bond. Walk along a helix:
N-Hi ... O=Ci+4,
N-Hi+1 ... O=Ci+5, …
Every N-H finds its partner four residues ahead in the
same chain. There are no inter-chain H-bonds in a free
α-helix; the entire stabilisation is internal.
Side chains stay out of the way. The R groups
project outward from the helix axis, so the H-bonded
backbone is shielded from solvent only by side chains. This
means the helix can sit on a protein's surface (with polar R
groups) or in the membrane (with hydrophobic R groups) without
losing its internal H-bond network.
Disrupting agents. Heat, urea, or detergents disrupt
the H-bonds and unwind the helix –- protein denaturation. The
peptide bonds (primary structure) survive because they are
covalent; only the secondary-structure H-bonds break.
α-Helix is stabilised by cooperative intra-chain N-H ⋯ O=C H-bonds, residue i to residue i+4.
Q 10.44
How do you explain the presence of five -OH groups in glucose molecule?
Concept used. Treatment of glucose with excess
acetic anhydride ((CH3CO)2O) in the presence of
a base such as pyridine yields glucose pentaacetate
(C6H7O(OCOCH3)5). Acetic anhydride esterifies every free
-OH. The formation of exactly five acetate esters
proves the presence of five free -OH groups.
Product analysis: 5 acetate groups have been installed.
Therefore glucose must have had five-OH groups.
Glucose forms a pentaacetate ⇒ 5 -OH groups.
DT
Dr. Tarun Kapoor
PhD Carbohydrate Chemistry, IIT Bombay
Verified Expert
Reaction in detail. Acetic anhydride reacts with free
hydroxyl groups under mildly basic conditions (pyridine soaks
up the acetic acid produced):
R-OH + (CH3CO)2O ->[pyridine] R-O-CO-CH3 + CH3COOH.
Each -OH on glucose is converted into an -O-COCH3
acetate ester. If glucose had, say, four -OHs, only a
tetraacetate would be possible. The clean isolation of a
pentaacetate (and not a hexa- or tetra-acetate)
proves there are exactly five free -OH groups.
Where the five -OH sit. In the open-chain
Fischer picture, glucose carries -OH at C-2, C-3, C-4,
C-5 and C-6. In the cyclic hemiacetal picture, four of these
remain (C-2, C-3, C-4, C-6) and a new hemiacetal -OH
appears at C-1 –- still five -OHs in total. Both
pictures are consistent with five acetates.
Cross-validation. The pentaacetate has a molecular
mass of 390. Combustion analysis,
hydrolysis (which regenerates 5 mol of acetic acid per mol of
pentaacetate) and NMR all confirm five -OCOCH3 groups.
The chemistry is unambiguous –- glucose has 5 -OHs.
Glucose forms a pentaacetate with acetic anhydride ⇒ five free -OH groups.
Q 10.45
Why does compound (A), glucose pentaacetate, not form an oxime?
Concept used. Oxime formation requires a free
carbonyl (C=O). In glucose pentaacetate the
cyclic hemiacetal-OH at C-1 has been converted
to an -OCOCH3 acetate ester, locking C-1 as a full
acetal. Acetals cannot ring-open back to the
-CHO form, so there is no free aldehyde available to
react with NH2OH⇒ no oxime.
Free glucose: cyclic hemiacetal C-1 -OH can open
to -CHO⇒ reacts with NH2OH.
Pentaacetate: C-1 -OH has become -OCOCH3⇒ now an acetal, not a hemiacetal.
Acetals are stable and do not regenerate -CHO
under mild conditions.
No free -CHO⇒ no oxime formation.
Pentaacetate's C-1 is an acetal, not a hemiacetal ⇒ no free -CHO⇒ no oxime.
LS
Lakshmi Subramanian
M.Sc Chemistry, IIT Madras
Verified Expert
Acetal vs hemiacetal distinction. A hemiacetal carbon
carries one -OR and one -OH (R–CH(OH)(OR'));
it can ring-open back to the parent aldehyde under aqueous or
acidic conditions. An acetal carbon carries two -OR groups
(R–CH(OR')(OR')); it is stable to water and does not
revert to the aldehyde without strong acid hydrolysis.
What acetylation does to C-1. In free glucose, the
cyclic form has C-1 bonded to a ring oxygen and a hemiacetal
-OH. Acetylation with (CH3CO)2O replaces the
-OH at C-1 with -OCOCH3. Now C-1 has two oxygen
substituents: the ring oxygen and the new acetate-ester oxygen.
Both are -OR-type oxygens ⇒ C-1 has become a
full acetal, locking the cyclic form.
No path to -CHO, no oxime. Oxime formation
requires nucleophilic addition of NH2OH to a C=O.
With C-1 locked as an acetal, the molecule cannot equilibrate
back to the open-chain aldehyde form. There is no C=O
available, so NH2OH has nothing to attack and no oxime
forms. Add a strong acid and the acetal will eventually
hydrolyse back to free glucose, after which NH2OH would
add successfully –- but under the standard mild oxime
conditions, pentaacetate is inert.
Pentaacetate locks C-1 as an acetal ⇒ no -CHO accessible ⇒NH2OH cannot form an oxime.
Q 10.46
Sucrose is dextrorotatory but the mixture obtained after hydrolysis is laevorotatory. Explain.
Concept used. Hydrolysis of sucrose gives equimolar
glucose + fructose. Pure sucrose has
[α]D = +66.5 (dextro). After hydrolysis,
glucose contributes +52.5 and fructose
-92.4. Because the magnitude of fructose's
laevorotation exceeds that of glucose's dextrorotation,
the net rotation of the mixture is negative
(∼ -20), i.e. laevorotatory.
Equimolar mix net rotation: +52.5 - 92.42 = -20 (laevo).
Net rotation flips + → - because fructose's laevorotation overrides glucose's dextrorotation. This mix is ``invert sugar''.
DS
Dr. Shreya Ghosh
PhD Chemistry, IIT Kharagpur
Verified Expert
Specific rotations to memorise.
[leftmargin=*,nosep]
Sucrose: [α]D = +66.5.
Glucose: [α]D = +52.5 at equilibrium.
Fructose: [α]D = -92.4.
Algebraic average. The optical rotation of an
equimolar mixture is the arithmetic mean of the two specific
rotations (since equal moles and equal molecular weights):
[α]mix
=
12(+52.5 + (-92.4))
=
-19.95 ≈ -20.
A clearly negative number ⇒ laevorotatory mixture.
Why the sign flips. The rotation of fructose is large
and negative; that of glucose is moderate and positive. When
combined 1:1, the negative term wins. The pre-hydrolysis
solution (sucrose alone) was strongly positive at
+66.5, so the change of sign is dramatic –-
hence the historical name ``invert sugar''.
Experimental observation. If you take a fresh sucrose
solution, add a drop of dilute HCl or some invertase, and
follow the polarimeter, you can watch the rotation drop from
+66.5 through zero down to -20
in real time –- a beautiful chemistry demonstration.
Sucrose's +66.5→ glucose (+52.5) + fructose (-92.4); mean is -20⇒ laevorotatory.
Q 10.47
Amino acids behave like salts rather than simple amines or carboxylic acids. Explain.
Concept used. An amino acid molecule contains both a
basic -NH2 and an acidic -COOH group on the same
carbon. In aqueous solution, an internal proton transfer
occurs: the -COOH donates H+ to the -NH2,
producing a zwitter ionH3N+-CHR-COO-. This dipolar ion is a salt-like species
that explains the high melting point, water solubility and
crystalline nature of amino acids.
In solid / solution: H2N-CHR-COOH -> H3N+-CHR-COO-
(zwitter ion).
The zwitter ion carries both positive (-NH3+) and
negative (-COO-) charges ⇒ behaves like
an ionic salt.
Hence high melting point (decomposition >200),
water solubility, low solubility in non-polar solvents
–- properties of salts, not of simple amines/acids.
Amino acids exist as zwitter ions (H3N+-CHR-COO-) ⇒ salt-like behaviour.
DK
Dr. Karan Malhotra
MBBS-MD Biochemistry, PGIMER Chandigarh
Verified Expert
Internal proton transfer. An amino acid in the dry
neutral form H2N-CHR-COOH is unstable because
-COOH (pKa ≈ 2–3) is a stronger acid than
-NH3+ is a strong acid (pKa ≈ 9–10). At
near-neutral pH the -COOH readily donates its proton to
the -NH2 on the same molecule:
H2N-CHR-COOH -> H3N+-CHR-COO-.
The result is a dipolar zwitter ion with both
positive and negative formal charges in the same molecule but
no net charge.
Salt-like consequences. Because the dominant form is
ionic, amino acids behave like inorganic salts rather than like
neutral molecules:
[leftmargin=*,nosep]
High melting points (>200,
often with decomposition) –- typical of ionic lattices.
Insoluble in non-polar solvents (ether,
chloroform) but soluble in water –- typical of
ionic compounds.
Migrate in an electric field –- behave like
cations at low pH and anions at high pH, with no
movement at the iso-electric point.
Do not show typical amine smell or acid
proton – amine and acid groups have neutralised each
other internally.
Experimental verification. The dipole moment of a
typical amino acid is ∼15D, far larger than the
∼2D of a neutral molecule of similar size –- direct
evidence of the separated +/- charges of the zwitter ion.
In water, amino acids exist as zwitter ions H3N+-CHR-COO-, so they behave like ionic salts (high m.p., water-soluble, dipolar).
Q 10.48
Structures of glycine and alanine are given below. Show the peptide linkage in glycylalanine. [2pt]
Glycine: H2N-CH2-COOH. Alanine: H2N-CH(CH3)-COOH.
Concept used. A peptide bond forms by condensation of
the -COOH of one amino acid with the -NH2 of the
next, with loss of H2O. The resulting linkage
-CO-NH- is a planar trans-amide.
In glycylalanine (Gly-Ala) the N-terminus is glycine
(its -NH2 stays free) and the C-terminus is
alanine (its -COOH stays free).
The -COOH of glycine condenses with the
-NH2 of alanine:
H2N-CH2-COOH + H2N-CH(CH3)-COOH -> H2N-CH2-CO-NH-CH(CH3)-COOH + H2O.
The bold -CO-NH- link in the centre is the
peptide bond.
Glycylalanine: H2N-CH2-CO-NH-CH(CH3)-COOH (peptide bond shown in bold).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Bond-by-bond view.
[leftmargin=*,nosep]
Start with glycine H2N-CH2-COOH and alanine
H2N-CH(CH3)-COOH.
Activate glycine's carboxyl carbon; align it with
alanine's amino nitrogen.
The amine nitrogen attacks the carboxyl carbon; a
tetrahedral intermediate forms; OH leaves
(as water, with the lost H coming from the
amine).
The result is the dipeptide
H2N-CH2-CO-NH-CH(CH3)-COOH with one water
molecule expelled.
Showing the linkage. The peptide bond is the
-CO-NH- link sitting between the α-C of glycine
and the α-C of alanine. We can also draw it more
explicitly:
H2N-CH2Gly residue
-
CO-NHpeptide bond
-
CH(CH3)-COOHAla residue.
Geometry note. The -CO-NH- unit is planar and
trans (the α-C of Gly and the α-C of Ala on
opposite sides of the C-N axis), because of partial double-bond
character from π-electron delocalisation between N and
C=O. This planar geometry is what allows the φ/ψ
dihedral angles of the backbone to take only certain values,
restricting the conformational space proteins can explore.
Glycylalanine: H2N-CH2-CO-NH-CH(CH3)-COOH; the central -CO-NH- is the peptide bond.
Q 10.49
A protein found in a biological system with a unique three-dimensional structure and biological activity is called a native protein. When a protein in its native form is subjected to a physical change (like change in temperature) or a chemical change (like change in pH), denaturation of protein takes place. Explain the cause.
Concept used. A protein's native 3-D shape is held by
non-covalent interactions –- H-bonds, hydrophobic
contacts, salt bridges –- and by occasional -S-S-
covalent bridges. Heat, extreme pH, urea, detergents or heavy
metals disrupt these interactions. The protein
unfolds: the α-helices uncoil, the β-sheets
unstack, and the globule disintegrates. The covalent peptide
bonds (primary structure) are not broken, but the
biological activity is lost because the precise 3-D geometry
needed for substrate binding is destroyed.
Loss of 2 and higher structures, but
peptide backbone (1) survives.
Hence denaturation: loss of biological activity
without breaking covalent peptide bonds.
Denaturation = disruption of non-covalent forces (H-bonds, hydrophobic, ionic) ⇒ unfolding ⇒ loss of biological activity. Peptide bonds survive.
AM
Aishwarya Menon
M.Sc Biochemistry, IISc Bangalore
Verified Expert
Forces that hold a native protein. The native fold is
built on non-covalent interactions:
[leftmargin=*,nosep]
Backbone H-bonds (the α-helix and β-sheet
scaffolding).
Side-chain H-bonds and salt bridges.
The hydrophobic effect (non-polar side chains huddle
inside, away from water).
Occasional -S-S- disulphide bridges (these are
covalent, but optional).
What denaturation actually breaks. Heat increases
kinetic energy and rattles H-bonds apart. Extreme pH
protonates or deprotonates side chains, destroying salt bridges
and altering local geometry. Urea or guanidinium chloride
out-competes the protein backbone for H-bonding water.
Detergents and organic solvents disrupt the hydrophobic core.
In each case, the non-covalent interactions fail; the
peptide backbone (a series of covalent -CO-NH- bonds)
survives.
What is lost. When the H-bond and hydrophobic networks
collapse, the helices uncoil and the sheets fall apart. The
3-D fold (tertiary structure) and the multi-chain assembly
(quaternary structure) disintegrate. The biological activity –-
which depends on a precise geometric arrangement of catalytic
residues or binding-site residues –- is destroyed. The
protein has been denatured.
Reversibility. Some small proteins (RNase A is the
classical example) refold spontaneously once the denaturant is
removed –- Anfinsen's experiment. Most large proteins (egg
albumin in a boiled egg) do not refold; the unfolded chains
get tangled with one another and precipitate.
Denaturation = breakdown of H-bonds, salt bridges, hydrophobic contacts under heat / pH change ⇒ unfolding of 2∘, 3∘, 4∘ structure ⇒ loss of biological activity. Primary structure survives.
Q 10.50
Activation energy for the acid catalysed hydrolysis of sucrose is 6.22, while the activation energy is only 2.15 when hydrolysis is catalysed by the enzyme sucrase. Explain.
Concept used. Enzymes are extraordinarily efficient
biocatalysts. They provide an alternative reaction
path with a lower activation energy (Ea) by binding the
substrate in a specific active site that lowers the
transition-state energy. The reaction rate is therefore
much higher than the corresponding acid-catalysed (or
uncatalysed) route at the same temperature.
Acid catalysis: Ea = 6.22 (path A).
Sucrase enzyme catalysis: Ea = 2.15 (path B).
Enzyme lowers Ea by Δ Ea = 6.22 - 2.15 = 4.07.
Rate constants ratio at T: kenz/kacid = exp(-Δ Ea/RT)
(Arrhenius) ⇒ at 310 K
exp(4070/(8.314 × 310)) ≈ exp(1.58) ≈ 4.85 times faster.
Hence sucrase hydrolyses sucrose much faster than H+ alone.
Sucrase provides a lower-Ea pathway (2.15 vs 6.22) ⇒ faster hydrolysis.
DS
Dr. Saurabh Joshi
PhD Enzymology, NCL Pune
Verified Expert
Arrhenius angle. The Arrhenius rate constant
k = Aexp(-Ea/RT) depends exponentially on Ea. A drop of
Δ Ea = 4.07 at body
temperature (T = 310,
RT = 2.58) gives a rate
enhancement of exp(4.07/2.58) ≈ exp(1.58) ≈ 4.85.
That alone explains the much faster sucrase-catalysed
hydrolysis in textbook calculations.
Why an enzyme can drop Ea so much. The enzyme
sucrase binds sucrose in its active site so tightly that the
substrate is already partly distorted toward the transition
state. Catalytic residues then accept a proton from the
glycosidic oxygen and donate one to the leaving group at the
right moment, breaking the C-O-C bond. Compared to a
random H3O+ collision in solution, this orchestrated
attack at the active site requires far less energy –- hence
the lower Ea.
Specificity bonus. Sucrase recognises only sucrose's
α,β-1,2 linkage. It does not hydrolyse maltose
(α-1,4) or lactose (β-1,4). Acid catalysis, in
contrast, hydrolyses any glycosidic bond non-selectively. So
the enzyme not only goes faster but also chooses its substrate
precisely –- two properties that make biology possible.
Enzyme sucrase lowers Ea from 6.22 to 2.15 by binding sucrose into a transition-state-like geometry ⇒ Arrhenius rate ∼ 5× faster.
Q 10.51
Which moieties of nucleosides are involved in the formation of phosphodiester linkages present in dinucleotides? What does the word ``diester'' in the name of the linkage indicate? Which acid is involved in the formation of this linkage?
Concept used. A phosphodiester linkage in a
polynucleotide bridges the 3'-OH of the sugar of
one nucleoside with the 5'-OH of the sugar of the
next, with one phosphate group esterified to both
hydroxyls –- hence ``di-ester''. The acid involved is
phosphoric acid (H3PO4).
Moieties involved: the 5'-OH and 3'-OH of the
pentose sugars of two adjacent nucleosides.
``Diester'' indicates that the central phosphorus carries
two-O-R ester bonds (one to each sugar).
Acid involved: phosphoric acid H3PO4.
5'-OH of one sugar + 3'-OH of the next, esterified twice by phosphoric acid ⇒ phosphodiester linkage.
KR
Kavita Reddy
M.Sc Molecular Biology, University of Hyderabad
Verified Expert
Naming the bond. The bridge that joins two nucleosides
in DNA / RNA is built from one phosphoric acid molecule
that has been esterified twice –- once at the 3'-OH of
sugar 1, and once at the 5'-OH of sugar 2. Because the same
phosphate forms two ester bonds, the whole linkage is called a
phosphodi-ester.
Sugar moieties involved. The pentose sugar of each
nucleoside contributes exactly one -OH to the linkage:
the 3'-OH on the upstream nucleoside, and the 5'-OH on the
downstream nucleoside. The base on C-1' is not involved in the
backbone linkage; it sits perpendicular to the chain and
participates in Watson–Crick pairing instead.
Acid involved. The bridging acid is
phosphoric acid, H3PO4 (or more accurately its
biological derivative, ATP/dNTPs, which donate the phosphate
during enzymatic polymerisation). Inside the chain the
phosphate retains one =O, one -OH (or -O-
at physiological pH), and two ester -O-R bonds.
Functional consequence. Because phosphate carries a
negative charge at physiological pH, the whole DNA backbone is
strongly anionic. This is why DNA migrates toward the positive
electrode in gel electrophoresis, and why histones (positively
charged proteins) are needed to package DNA in chromosomes.
Sugar moieties: 3'-OH of one nucleoside + 5'-OH of the next; ``diester'' = two ester bonds on one phosphate; acid = phosphoric acid H3PO4.
Q 10.52
What are glycosidic linkages? In which type of biomolecules are they present?
Concept used. A glycosidic linkage is a
C-O-C ether bond that joins two monosaccharide units
through the anomeric carbon of one sugar and a hydroxyl carbon
of the next, with elimination of one water molecule. It is
the standard inter-monomer bond in all di-, oligo-
and polysaccharides (carbohydrates).
Glycosidic linkage = C-O-C ether between sugar units; present in carbohydrates (di-/oligo-/polysaccharides).
NV
Nisha Verma
JEE Faculty, Resonance Kota
Verified Expert
Formation step. A glycosidic linkage is formed when
the hemiacetal (or hemiketal) -OH on the anomeric C of
one sugar attacks any -OH of a neighbouring sugar; one
molecule of water leaves and a C-O-C ether bridge
remains. The bridge is named by the two carbons it joins (e.g.
``1,4'' or ``1,6'') and by the stereochemistry of the
anomeric C (α or β).
α-1,6: branch points in amylopectin and glycogen;
isomaltose.
α,β-1,2: sucrose (both anomeric C used).
Biomolecule class. Glycosidic linkages live almost
exclusively in carbohydrates (disaccharides such as maltose,
sucrose, lactose; oligosaccharides such as raffinose;
polysaccharides such as starch, cellulose, glycogen, chitin).
A related class –- the N-glycosidic linkage –- also joins the
anomeric C to a base nitrogen in nucleosides (e.g. adenine to
ribose to make adenosine).
Glycosidic linkages are C-O-C bonds joining sugar units; they are the backbone of carbohydrates (di-, oligo- and polysaccharides).
Q 10.53
Which monosaccharide units are present in starch, cellulose and glycogen, and which linkages link these units?
Concept used. All three polysaccharides are polymers of
D-glucose, but their glycosidic linkages differ in
stereochemistry and branching, which produces dramatically
different biological roles.
Starch: polymer of α-D-glucose;
amylose component is linear α-1,4;
amylopectin component is α-1,4 + occasional
α-1,6 (branch points every ∼ 25 residues).
Cellulose: polymer of β-D-glucose; linear
β-1,4 only; no branching.
Glycogen: polymer of α-D-glucose;
α-1,4 chain plus frequent α-1,6 branches
(every ∼ 10 residues, denser than amylopectin).
Common monomer, different bonds. Starch, cellulose and
glycogen are all polysaccharides built from the single
monosaccharide D-glucose. What distinguishes them is the
stereochemistry of the glycosidic bond and the
branching topology.
Starch. The major plant storage polysaccharide is a
mixture of two components:
[leftmargin=*,nosep]
Amylose (∼ 20%): linear chains of α-D-glucose
joined by α-1,4 glycosidic bonds. Helical coil.
Amylopectin (∼ 80%): branched chains of
α-D-glucose with α-1,4 main chain and
α-1,6 branch points every ∼ 25 residues.
Cellulose. The major structural polysaccharide of plant
cell walls. Linear chains of β-D-glucose joined by
β-1,4 bonds. No branching. The chains lie flat and
H-bond into microfibrils, giving wood its rigidity and cotton
its tensile strength.
Glycogen. The animal storage polysaccharide, found
mainly in liver and skeletal muscle. Identical bond pattern to
amylopectin –- α-1,4 main chain plus α-1,6
branches –- but with branches every ∼ 8–12 residues
(denser than amylopectin). The denser branching gives more
non-reducing ends for rapid mobilisation by glycogen
phosphorylase.
All three are D-glucose polymers; α-linkages give starch/glycogen (digestible storage), β-linkage gives cellulose (structural fibre).
Q 10.54
How do enzymes help a substrate to be attacked by the reagent effectively?
Concept used. Enzymes have a specific
active site –- a small pocket on the protein surface
shaped to fit a particular substrate by complementary geometry
and chemistry (``lock-and-key'' or ``induced fit''). When the
substrate binds, it is held in a precise orientation that
exposes the reactive bond to the catalytic residues / co-factors
that act as the ``reagent''. The result is a much faster
reaction at a much lower activation energy.
Substrate diffuses into the enzyme's active site and is
held by H-bonds, salt bridges, hydrophobic contacts.
The active site geometry forces the reactive bond into a
strained, transition-state-like conformation.
Catalytic residues (His, Asp, Glu, Ser, Cys) and/or
cofactors (NAD+, FAD, metal ions) attack the substrate
from precisely the right angle.
Reaction proceeds rapidly; product diffuses out and the
active site resets.
Active site binds substrate in the right orientation and brings catalytic groups close ⇒ effective attack and low Ea.
RA
Ritu Agarwal
NEET Educator, Vedantu
Verified Expert
Active-site mechanics. An enzyme's active site is a
small cleft on the protein surface, shaped through the protein's
3-D fold to be a perfect complement to its substrate. When a
substrate molecule diffuses in:
[leftmargin=*,nosep]
It is gripped by an array of non-covalent interactions
(H-bonds, ionic interactions, hydrophobic contacts)
between the substrate functional groups and the
side-chains of the enzyme.
These interactions hold the substrate in a precise
position, often distorted toward the transition-state
geometry (this is the essence of ``induced fit'' /
transition-state stabilisation).
Specific amino-acid residues in the active site
(catalytic residues like serine, histidine, aspartate,
cysteine) sit at exactly the right distance and angle
to attack the reactive bond, either donating/accepting
protons, or providing a nucleophile.
In many enzymes a cofactor (NAD+, FAD, Zn2+,
haem) is bound at the active site and contributes the
actual reactive species.
Net effect. The substrate is no longer at the mercy of
random thermal collisions with a free reagent; it is locked in
position, oriented favourably, and reacts much faster. This is
why enzymes can speed up reactions by factors of 106 to
1017 at body temperature, where no industrial catalyst
matches them.
Lock-and-key / induced-fit picture. Emil Fischer
proposed the rigid ``lock-and-key'' model. Daniel Koshland
refined it to ``induced fit'': the enzyme is somewhat flexible,
and on binding the substrate it deforms slightly to embrace the
substrate more tightly. Both pictures explain how the substrate
ends up in the perfect position for catalytic attack.
Enzyme active site grips the substrate in the right orientation; catalytic side-chains/cofactors then attack effectively ⇒ low Ea, high rate, high specificity.
Q 10.55
Describe the term D- and L-configuration used for amino acids with examples.
Concept used. An amino acid is assigned the
D or L label by comparing the orientation
of its -NH2 on the α-carbon (Fischer projection,
-COOH at top) with that of D- or L-glyceraldehyde.
-NH2 on the right → D; on the left → L.
Draw the amino acid in Fischer projection: -COOH
at the top, -R (side chain) at the bottom.
Locate the α-carbon (the one bearing -NH2,
-H and -R).
If -NH2 is on the right ⇒ D; on the
left ⇒ L.
Examples.
[leftmargin=*,nosep]
L-alanine: -NH2 on left of α-C (natural form).
D-alanine: -NH2 on right of α-C (in bacterial cell walls).
L-glutamic acid, L-lysine, L-phenylalanine: all natural
protein-forming amino acids are L-configured.
D = -NH2 right of α-C; L = -NH2 left of α-C. Almost all proteinogenic amino acids are L.
DA
Dr. Amit Banerjee
PhD Biochemistry, Bose Institute Kolkata
Verified Expert
Origin of the labels. The D/L notation was introduced
by Emil Fischer to relate the configuration of any
chiral molecule to that of D- or L-glyceraldehyde, the
simplest chiral aldose. For an amino acid, place the molecule
in Fischer projection with -COOH at the top, side chain
-R at the bottom, and look at the position of -NH2
on the α-carbon (the carbon adjacent to -COOH).
If -NH2 sits on the right, the amino acid is
D-configured; if on the left, L-configured.
Examples in detail.
[leftmargin=*,nosep]
L-Alanine: Fischer projection shows
-COOH on top, -H on right of α-C,
-NH2 on left, -CH3 at bottom. This is the
form used in all natural proteins.
D-Alanine: mirror image of L-alanine with
-NH2 on the right. Found in the peptidoglycan of
bacterial cell walls; substrate of bacterial racemases.
L-Glutamic acid: a five-carbon acidic amino
acid; Fischer-left -NH2 on α-C; the form
responsible for umami taste in MSG.
D vs L vs (+)/(-). As with sugars, the D/L label
captures configuration, not the experimental optical
rotation. L-alanine, for instance, has
[α]D = +14.5 in 5N HCl –- it is
dextrorotatory despite being L-configured. So always
read the prefix capital ``L'' as a structural label, not as a
direction of rotation.
L = -NH2 left of α-C in Fischer; D = right. All natural protein amino acids are L. Examples: L-Ala, L-Glu, L-Lys.
Q 10.56
How will you distinguish 1∘ and 2∘ hydroxyl groups present in glucose? Explain with reactions.
Concept used. Glucose has one primary
-OH (at C-6, on -CH2OH) and four
secondary -OHs (at C-2, C-3, C-4, C-5).
Conc. HNO3 oxidises the C-6 -CH2OHonly (along with the C-1 -CHO) to give a
dicarboxylic acid (saccharic acid). Secondary
hydroxyls are not oxidised further under these conditions, so
they remain as -CHOH-. The very formation of a
dicarboxylic acid is the test for the lone primary -OH.
Both terminal C's become -COOH⇒ C-1
-CHO and C-6 -CH2OH are at terminal
positions and the four secondary -CHOHs in the
middle remain unchanged.
Hence primary-OH at C-6 oxidises to
-COOH; secondary-OHs at C-2–C-5
survive ⇒ distinguishing test is dicarboxylic
acid formation.
Conc. HNO3 oxidises only the primary C-6 -CH2OH (and the C-1 -CHO) of glucose to give saccharic acid; the 4 secondary -OHs survive.
DT
Dr. Tarun Kapoor
PhD Carbohydrate Chemistry, IIT Bombay
Verified Expert
Distinguishing the two types of hydroxyl. Glucose has
six carbons. C-1 is an aldehyde, C-6 is a primary alcohol
(-CH2OH), and C-2, C-3, C-4, C-5 are secondary alcohols
(-CHOH-, each carbon bearing two non-hydrogen
substituents). The chemistry test that pinpoints the primary
alcohol exploits the fact that primary -OHs oxidise to
-COOH under strongly oxidising conditions, whereas
secondary -OHs either go to ketones (with mild
oxidants) or get protected (with strong HNO3 they are
mostly inert).
Two-step oxidation experiment.
[leftmargin=*,nosep]
Treat glucose with bromine water
(Br2/H2O, mild, room temperature). The only
functional group that oxidises is the aldehyde at C-1
→-COOH. Product: gluconic acidHOCH2-(CHOH)4-COOH, a monocarboxylic acid.
This confirms an aldehyde but not the primary
-OH – Br2/H2O is too mild for that.
Treat glucose with conc. HNO3 with heat.
Both terminal carbons get oxidised: C-1 -CHO→-COOH (as before) and C-6
-CH2OH→-COOH. Product:
saccharic acid (glucaric acid)HOOC-(CHOH)4-COOH –- a dicarboxylic acid.
The four middle -OHs (C-2, C-3, C-4, C-5)
remain unoxidised in saccharic acid.
Diagnostic reasoning. The contrast between the
monocarboxylic gluconic acid and the dicarboxylic saccharic
acid is the proof that C-6 is a primary -OH (oxidisable
to -COOH) and C-2 to C-5 are secondary -OHs
(not oxidised further under these conditions). Hence the
primary and secondary hydroxyls of glucose are
distinguished.
Conc. HNO3 converts the primary C-6 -CH2OH and the C-1 -CHO to -COOH (saccharic acid); the four secondary -OHs on C-2 to C-5 are unaffected –- the dicarboxylic-acid test distinguishes 1∘ from 2∘ hydroxyls.
Q 10.57
Coagulation of egg white on boiling is an example of denaturation of protein. Explain it in terms of structural changes.
Concept used. Egg white is mostly the globular protein
albumin, with its native 3-D fold held by H-bonds,
hydrophobic interactions, salt bridges and a few disulphide
bridges. Boiling supplies enough thermal energy to break the
non-covalent interactions. The compact globular protein
unfolds: α-helices uncoil, β-sheets
unstack, the hydrophobic core is exposed, and hydrophobic
patches on different unfolded chains aggregate. The aggregate
is the white, opaque, insoluble coagulum we see.
Exposed hydrophobic patches on different chains
aggregate (4 aggregation), trapping water in
a gel and scattering light ⇒ opaque white.
Peptide bonds (1 structure) are unaffected
⇒ no peptide hydrolysis.
Boiling disrupts H-bonds / hydrophobic / ionic interactions of egg albumin ⇒ unfolding and aggregation ⇒ coagulation; peptide bonds survive.
AK
Anjali Krishnan
NEET Faculty, Allen Kota
Verified Expert
Native albumin. Egg white (the ``albumen'') is about
10% protein by mass; the dominant component is the
45 globular protein albumin (ovalbumin).
In its native state the chain folds into a compact, roughly
spherical globule with the hydrophobic residues buried in the
core and the polar residues on the surface. H-bonds (helix and
sheet), hydrophobic contacts (core), salt bridges
(Lys-NH3+⋯Glu-COO-) and a small number of
disulphide bridges keep the fold intact.
Effect of boiling. At 100 the
average kinetic energy per molecule
(∼ kBT ≈ 3 per degree
of freedom) is enough to rattle apart the H-bonds and disrupt
the salt bridges; the hydrophobic interactions also weaken at
high temperature. The chain unfolds: α-helices uncoil,
β-sheets unstack, the globule disintegrates. The previously
buried hydrophobic residues are now exposed to water –- a
thermodynamically unfavourable state.
Aggregation → coagulation. The exposed hydrophobic
patches on different unfolded chains stick to each other to
escape water (the hydrophobic effect again, but now between
chains). The chains tangle into a three-dimensional polymer
network that traps water inside, forming a gel. The network is
large enough to scatter visible light, so the previously
transparent egg-white becomes opaque white. Once the network
forms, the tangled chains cannot find their way back to the
native fold –- the coagulation is irreversible at the
macroscopic scale.
What does not happen. The peptide bonds
-CO-NH- are covalent and survive boiling completely.
The primary structure (sequence of amino acids) is unchanged.
What is lost is the 2 (α-helix, β-sheet),
3 (tertiary fold) and 4 (oligomeric
state) structure –- and with them, the biological activity.
Boiling denatures egg-albumin: H-bonds and hydrophobic interactions break, the globule unfolds, hydrophobic patches aggregate into a gel-like network ⇒ opaque white coagulum. Peptide bonds (1) survive.
IV. Matching Type
Q 10.58
Match the vitamins given in Column I with the deficiency disease they cause given in Column II. [2pt]
tabularp0.42p0.45
Column I (Vitamins) & Column II (Diseases)
(i) Vitamin A & (a) Pernicious anaemia
(ii) Vitamin B1 & (b) Increased blood clotting time
(iii) Vitamin B12 & (c) Xerophthalmia
(iv) Vitamin C & (d) Rickets
(v) Vitamin D & (e) Muscular weakness
(vi) Vitamin E & (f) Night blindness
(vii) Vitamin K & (g) Beri Beri
& (h) Bleeding gums
& (i) Osteomalacia
tabular
Concept used. Each vitamin participates in a specific
biochemical pathway; deficiency disrupts that pathway and
produces a characteristic disease. Note that vitamins A and D
each cause two listed diseases (multiple matches are allowed).
Vitamin A → retinal pigment for vision & mucous-membrane
integrity ⇒ deficiency causes
xerophthalmia (c) and night blindness (f).
Vitamin B1 (thiamine) → cofactor for pyruvate
decarboxylation ⇒ deficiency causes beri
beri (g).
Biochemical-cause angle. Each vitamin acts as a cofactor
for a precise biochemical step. Knock that step out and the
disease appears. Walk through each vitamin:
[leftmargin=*,nosep]
Vitamin A (retinol) → retinal pigment in rod cells;
also keeps mucous-membrane / cornea hydrated ⇒
deficiency → night blindness (f) and
xerophthalmia (c).
Vitamin B1 (thiamine) → thiamine pyrophosphate
is the coenzyme for pyruvate dehydrogenase; without it
nervous tissue starves ⇒ beri-beri (g).
Vitamin B12 (cobalamin) → needed by
methionine synthase and methylmalonyl-CoA mutase;
deficiency → defective DNA synthesis in
erythroblasts ⇒ pernicious anaemia (a).
Vitamin C (ascorbic acid) → cofactor for
prolyl/lysyl hydroxylase in collagen synthesis;
deficiency → scurvy with bleeding gums (h).
Vitamin D (calcitriol) → regulates intestinal
Ca2+ absorption; deficiency in children →
rickets (d), in adults → osteomalacia (i).
Vitamin E (tocopherol) → lipid antioxidant for
membrane integrity; deficiency → muscular weakness
and haemolytic anaemia (e).
Vitamin K →γ-carboxylates glutamate
residues on prothrombin and other clotting factors;
deficiency → increased clotting time (b).
Final pairing.
(i) A→(c), (f),
(ii) B1→(g),
(iii) B12→(a),
(iv) C→(h),
(v) D→(d), (i),
(vi) E→(e),
(vii) K→(b).
Two vitamins (A and D) match two diseases each –- typical
of matching items where the answer key is many-to-one or
many-to-many. Always read the instruction line to confirm whether
multiple matches are permitted.
Each vitamin pairs with the specific disease produced by losing its cofactor function in metabolism.
Q 10.59
Match the following enzymes given in Column I with the reactions they catalyse given in Column II. [2pt]
tabularp0.42p0.45
Column I (Enzymes) & Column II (Reactions)
(i) Invertase & (a) Decomposition of urea into NH3 and CO2
(ii) Maltase & (b) Conversion of glucose into ethyl alcohol
(iii) Pepsin & (c) Hydrolysis of maltose into glucose
(iv) Urease & (d) Hydrolysis of cane sugar
(v) Zymase & (e) Hydrolysis of proteins into peptides
tabular
Concept used. Each enzyme is named for the substrate it
attacks (substrate-ase) or for the broad class of reactions
it catalyses. Matching is therefore a direct application of
enzyme-substrate pairing.
Maltase → hydrolyses maltose into 2 glucose
⇒ (c).
Pepsin → stomach protease, hydrolyses proteins to
peptides ⇒ (e).
Urease → hydrolyses urea to NH3 + CO2⇒ (a).
Zymase → yeast enzyme that ferments glucose to ethanol
⇒ (b).
(i)→(d); (ii)→(c); (iii)→(e); (iv)→(a); (v)→(b).
DS
Dr. Saurabh Joshi
PhD Enzymology, NCL Pune
Verified Expert
Walk-through.
[leftmargin=*,nosep]
Invertase (also called sucrase) cleaves the
α,β-1,2 glycosidic bond of sucrose (cane sugar)
into glucose + fructose. So invertase → (d) hydrolysis
of cane sugar.
Maltase cleaves the α-1,4 bond of maltose
into two glucose molecules. So maltase → (c)
hydrolysis of maltose into glucose.
Pepsin is a stomach protease secreted as
inactive pepsinogen and activated by gastric HCl
(pH≈ 2). It hydrolyses peptide bonds
in dietary proteins, especially at Phe/Tyr/Leu/Trp
residues, producing shorter peptides. So pepsin →
(e) hydrolysis of proteins to peptides.
Urease is a nickel-containing enzyme (first
enzyme ever crystallised, by Sumner in 1926) that
hydrolyses urea
urea (H2N)2C=O + H2O yields 2 NH3 + CO2 (catalyst: urease). So
urease → (a) decomposition of urea into NH3
and CO2.
Zymase is the complex of yeast enzymes
(including pyruvate decarboxylase and alcohol
dehydrogenase) that converts glucose to ethanol and
CO2 during fermentation:
C6H12O6 ->[zymase] 2 C2H5OH + 2 CO2. So zymase
→ (b) conversion of glucose to ethyl alcohol.
Assertion (A): D-(+)-Glucose is dextrorotatory in nature. Reason (R): `D' represents its dextrorotatory nature.
Correct option: (iii) A is correct, R is wrong.
Concept used. The prefix D/L describes
configuration (the orientation of -OH on the
lowest chiral carbon, comparing to D- or L-glyceraldehyde).
The signs (+)/(-) (or d/l) describe the
direction of optical rotation as measured by a
polarimeter –- this is an experimental fact, unrelated to the
D/L label.
Verify A: glucose rotates plane-polarised light to the
right ⇒ ``+'' or dextrorotatory.
A is true.
Examine R: D simply means the -OH on C5 is on the
right in Fischer projection (D-configuration). The
D label does not guarantee dextrorotation; e.g.
D-fructose is laevorotatory (-92.4∘). R is false.
Therefore A true, R false ⇒ option (iii).
D = configuration only; (+) = experimental rotation.
DS
Dr. Shreya Ghosh
PhD Chemistry, IIT Kharagpur
Verified Expert
Two distinct concepts. The capital prefix ``D'' is a
configurational descriptor based on Fischer projection
geometry –- it tells you that the chiral -OH on the
lowest stereocentre (C-5 in glucose) sits on the right
side, just as in D-glyceraldehyde. It says nothing about how the
molecule rotates plane-polarised light. The sign (+) or (-)
is an experimental observable measured in a polarimeter;
+ means dextrorotatory, - means laevorotatory.
Test A. D-(+)-Glucose really does rotate
plane-polarised light to the right by +52.5
(specific rotation, 20∘C, sodium D-line). So Assertion is
true.
Test R. The reason claims that the letter ``D'' itself
represents dextrorotation. That is wrong. D is a configurational
label only; the rotation sign is independent. Concrete
counter-example: D-(-)-fructose rotates the plane to
the left (-92.4), even though it is still
a D-sugar. So Reason is false.
Verdict. A true, R false ⇒ option (iii). The
trap here is that ``D'' and ``d'' (or (+) ) share the same
opening letter, tempting students to conflate them. The
historical naming is also a trap: Emil Fischer originally
thought that all D-sugars rotated right, but later
discoveries (D-fructose) overturned that link.
D ≡ Fischer configuration; (+)≡ optical rotation. Independent labels.
Q 10.61
Assertion (A): Vitamin D can be stored in our body. Reason (R): Vitamin D is fat soluble vitamin.
Correct option: (i) Both A and R are correct and R
explains A.
Concept used.Fat-soluble vitamins
(A, D, E, K) dissolve in body fat and are stored in adipose
tissue and the liver, so a daily intake is not strictly needed.
Water-soluble vitamins (B-complex, C) cannot be stored
and any excess is excreted in urine.
Vitamin D is one of the fat-soluble vitamins (A, D, E, K)
⇒ R is true.
Fat-soluble vitamins dissolve in body fat and accumulate
in liver/adipose tissue ⇒ they can be
stored⇒ A is true.
R is exactly the mechanistic reason why vitamin D can be
stored ⇒ R explains A.
Both correct, R explains A ⇒ option (i).
Vitamin D is fat-soluble ⇒ stored in body ⇒ (i).
DM
Dr. Manoj Tiwari
MBBS-MD, KGMU Lucknow
Verified Expert
Independent verification of A. Vitamin D is taken up
along with dietary fats in chylomicrons, delivered to the
liver, and then deposited in body fat. The liver and adipose
tissue act as reservoirs that can last weeks to months, even
without further dietary intake. So Assertion –- ``vitamin D can
be stored in our body'' –- is true.
Independent verification of R. Vitamin D
(cholecalciferol, C27H44O) is a derivative of cholesterol;
it is a non-polar steroid with one -OH group –- the
hydroxyl is too small to overcome the bulk of the steroid
nucleus, so the molecule remains highly fat-soluble. So Reason
–- ``vitamin D is a fat-soluble vitamin'' –- is also true.
Causal link between A and R. The very property of being
fat-soluble is what enables storage. Fat-soluble
molecules partition into adipose triglycerides; water-soluble
molecules cannot. Therefore R is the mechanistic explanation of
A, not just a correlated fact. The verdict is option (i): both
correct, and R explains A.
Beyond the question –- toxicity. Because vitamin D is
stored, mega-dose supplementation
(>100) can produce hypervitaminosis-D
–- hypercalcaemia, kidney stones, soft-tissue calcification.
This is impossible with water-soluble vitamins because the body
excretes excess in urine within hours.
Vitamin D is fat-soluble ⇒ stored in adipose tissue; both A and R true, R causes A ⇒ (i).
Q 10.62
Assertion (A):β-glycosidic linkage is present in maltose. Reason (R): Maltose is composed of two glucose units in which C-1 of one glucose unit is linked to C-4 of another glucose unit.
Correct option: (iv) A is wrong, R is correct.
Concept used. Maltose is a disaccharide of two
α-D-glucose units linked by an α-1,4
glycosidic bond, not a β-1,4 bond. The Reason
correctly states the C1–C4 connection. The Assertion is wrong
because it claims a β-linkage where actually the linkage
is α.
Check A: maltose has α-1,4 glycosidic linkage,
not β-1,4 ⇒ A is false.
Check R: yes, C-1 of one glucose is linked to C-4 of the
other ⇒ R is true.
A false, R true ⇒ option (iv).
Maltose has α-1,4 linkage; A wrong, R correct ⇒ (iv).
SP
Sneha Pillai
NEET Educator, Unacademy
Verified Expert
Verify A. Maltose is built from two α-D-glucose
units. The anomeric carbon of the first glucose (C-1) is
in the α-configuration (OH below the ring plane). It is
this α-OH that condenses with the C-4 OH of the second
glucose to form the glycosidic bond. So the linkage is
α-1,4, not β-1,4. The Assertion is
therefore false.
Verify R. The Reason correctly identifies that maltose
is two glucoses joined by a C-1 to C-4 bond. So the position of
the link is right; only the stereochemistry (α vs β)
was misstated in A. The Reason is therefore true.
Verdict. A is false but R is true ⇒ option
(iv) in the A-R schema.
Why we care about α vs β. An α-1,4
bond is what mammalian salivary and pancreatic amylases can
hydrolyse ⇒ we can digest maltose (and starch). A
β-1,4 bond requires cellulase, which mammals lack
⇒ we cannot digest cellulose. So the difference of
one letter (α vs β) has huge dietary consequences.
Maltose has α-1,4 glycosidic linkage (not β). A false, R true ⇒ option (iv).
Q 10.63
Assertion (A): All naturally occurring α-amino acids except glycine are optically active. Reason (R): Most naturally occurring amino acids have L-configuration.
Correct option: (v) Both A and R are correct but R does not explain A.
Concept used. Optical activity requires a
chiral carbon (4 different substituents). Every
α-amino acid except glycine has a chiral α-C
(four substituents: -COOH, -NH2, -H, -R),
hence optically active. The L-configuration of natural amino
acids is a separate, additional fact –- but it is the chirality
of the α-C (not the choice of L or D) that causes optical
activity.
Check A: chirality requires 4 different groups; glycine
has -R = -H⇒ two identical groups
⇒ not chiral; all other amino acids are chiral
⇒ A true.
Check R: naturally occurring proteinogenic amino acids
are L ⇒ R true.
Explanation link: optical activity follows from chirality,
not from D/L (a D-amino acid would also be optically
active). So R does not explain A.
Both A and R correct but R doesn't explain A ⇒ option (v).
A and R both true but unrelated; chirality (not L-configuration) causes optical activity ⇒ option (v).
DS
Dr. Shreya Ghosh
PhD Chemistry, IIT Kharagpur
Verified Expert
Logic of A. An α-amino acid has the general
formula H2N-CHR-COOH. Its α-C bears four
substituents: -COOH, -NH2, -H, and
-R. If these four are all different, the α-C is a
chirality centre and the molecule is optically active. For
glycine (R = -H), two of the four substituents are
-H⇒ not chiral ⇒ not optically
active. For every other proteinogenic amino acid,
R ≠ -H, so the α-C is chiral ⇒
optically active. The Assertion is true.
Logic of R. The Reason states that natural amino acids
have L-configuration. This is true: ribosomes assemble only L-
amino acids, so all proteinogenic amino acids in living cells
are L.
Why R does not explain A. Optical activity arises from
chirality of the α-C, which depends only on the
presence of four different substituents –- not on
which enantiomer (L or D) the cell happens to use. A
hypothetical organism using D-amino acids would still have
optically active amino acids. So R is a true but logically
independent statement; it doesn't cause A. Hence the
correct A-R option is (v): both correct, but R does not
explain A.
Both A and R true; but chirality (4 different groups), not L-configuration, is what makes amino acids optically active ⇒ option (v).
Q 10.64
Assertion (A): Deoxyribose, C5H10O4, is not a carbohydrate. Reason (R): Carbohydrates are hydrates of carbon, so compounds which follow the formula Cx(H2O)y are carbohydrates.
Correct option: (ii) Both A and R are wrong statements.
Concept used. The modern definition of carbohydrate is
polyhydroxy aldehyde or ketone, or a compound that
hydrolyses to such a unit. The historical name comes from
Cx(H2O)y, but many compounds with that formula are
not carbohydrates (e.g. formaldehyde CH2O),
and many genuine carbohydrates (e.g. deoxyribose
C5H10O4, rhamnose C6H12O5) do not follow
the empirical Cx(H2O)y ratio. Hence the modern
chemistry definition supersedes the old etymological one.
Check A: deoxyribose is an aldopentose (pentose with
-H in place of one -OH at C-2). It is a
polyhydroxy aldehyde and undergoes typical sugar
chemistry ⇒ it is a carbohydrate
⇒ A is false.
Check R: the Cx(H2O)y ``hydrate of carbon''
criterion is historical and outdated; many carbohydrates
don't fit (deoxy sugars), and many non-carbohydrates
do (formaldehyde, acetic acid C2H4O2). Hence the
formula is not a valid definition ⇒ R is false.
Both A and R false ⇒ option (ii).
Deoxyribose IS a carbohydrate; Cx(H2O)y is not the modern definition. Both A and R wrong ⇒ (ii).
DV
Dr. Vikram Saini
PhD Organic Chemistry, IISc Bangalore
Verified Expert
Why A is false. Deoxyribose (C5H10O4) is a
polyhydroxy aldehyde: HOCH2-CHOH-CHOH-CH2-CHO (in open
chain form) with a -CHO at C-1, -CH2- at C-2 (the
``deoxy'' position), and three -OHs on C-3, C-4 and C-5
(written here with C-1 on the right by the Fischer convention,
but the key fact is the aldehyde and three hydroxyls). It
undergoes every typical sugar reaction –- forms a furanose
hemiacetal, reduces Tollens' reagent, reacts with phenylhydrazine
to give an osazone, etc. By the modern chemistry definition, it
is unambiguously a carbohydrate (specifically, an aldopentose).
So the Assertion ``deoxyribose is not a carbohydrate'' is
false.
Why R is false. The Reason restates the historical
etymology –- the word ``carbohydrate'' was coined when
chemists noticed that many sugars had the empirical formula
Cx(H2O)y (e.g. glucose C6H12O6 = (CH2O)6).
But this empirical relationship is neither necessary nor
sufficient to define a carbohydrate:
[leftmargin=*,nosep]
Counterexample 1: formaldehyde HCHO = CH2O
fits the formula but is not a carbohydrate.
Counterexample 2: acetic acid C2H4O2 = C2(H2O)2
fits the formula but is not a carbohydrate.
Counterexample 3: deoxyribose C5H10O4is a
carbohydrate but does NOT fit Cx(H2O)y
(it would need C5H10O5). Similarly rhamnose
C6H12O5.
So the ``hydrate of carbon'' rule is not a valid definition.
The Reason is therefore false too.
Modern definition. A carbohydrate is a polyhydroxy
aldehyde or ketone, or a compound that hydrolyses to such a
unit. Deoxyribose satisfies this definition (aldopentose with
3 hydroxyls) ⇒ it is a carbohydrate.
Both A and R are false (deoxyribose IS a carbohydrate; Cx(H2O)y is not the defining criterion). Option (ii).
Q 10.65
Assertion (A): Glycine must be taken through diet. Reason (R): It is an essential amino acid.
Correct option: (ii) Both A and R are wrong.
Concept used.Essential amino acids are those
the human body cannot synthesise and that must be supplied by
diet. Glycine, however, is a non-essential amino acid
–- the body synthesises it from serine (via serine
hydroxymethyltransferase) and from threonine. So neither does
glycine need to come from diet, nor is glycine an essential
amino acid.
Check A: glycine is synthesised endogenously (mainly
from serine) ⇒ does not need to come
from diet ⇒ A is false.
Check R: glycine is non-essential ⇒ R is false.
Both A and R false ⇒ option (ii).
Glycine is non-essential (the body makes it from serine). Both A and R false ⇒ option (ii).
DN
Dr. Naveen Khanna
MBBS, Maulana Azad Medical College
Verified Expert
Essential vs non-essential. ``Essential'' in nutrition
specifically means ``cannot be synthesised by the body and
therefore must come from diet''. The nine essential amino acids
for adult humans are phenylalanine, valine, threonine,
tryptophan, isoleucine, methionine, leucine, lysine, histidine.
(Arginine is sometimes called the tenth, conditionally
essential.) Glycine is not on this list.
Glycine biosynthesis. The body synthesises glycine via
several routes:
[leftmargin=*,nosep]
From serine via serine hydroxymethyltransferase
(the major route): serine → glycine + -CH2-
(transferred to tetrahydrofolate).
From threonine via threonine dehydrogenase.
From the glyoxylate pathway (alanine + glyoxylate
→ glycine + pyruvate via the glyoxylate
aminotransferase).
Because of these routes, glycine is classified as
non-essential. A is therefore false.
R is also false. Glycine is not an essential amino
acid. Therefore the Reason itself is incorrect, independent of
its supposed link to A. Both A and R are false statements
⇒ option (ii).
Edge case. Under certain stress conditions (premature
infants, recovery from major surgery, burns), glycine demand
may outpace synthesis temporarily and glycine becomes
``conditionally essential''. But in a normal adult, glycine
is non-essential.
Glycine is non-essential; the body makes it from serine. Both A and R false ⇒ option (ii).
Q 10.66
Assertion (A): In presence of an enzyme, substrate molecules can be attacked by the reagent effectively. Reason (R): Active sites of enzymes hold the substrate molecule in a suitable position.
Correct option: (i) Both A and R are correct, and R explains A.
Concept used. An enzyme's active site grips
the substrate in a precise orientation (induced fit). With the
substrate locked into a transition-state-like geometry,
catalytic residues or co-factors can attack the reactive bond
from the optimum angle. Hence the effective attack of the
substrate is a direct consequence of the active-site
positioning.
Check A: enzymes accelerate reactions by orders of
magnitude ⇒ A is true.
Check R: active sites hold the substrate in a specific
orientation by complementary geometry and chemistry
⇒ R is true.
Link: holding the substrate in the right pose is
precisely why the reagent attacks effectively
⇒ R explains A.
Both correct, R explains A ⇒ option (i).
Active-site positioning of substrate is the reason for effective enzyme attack ⇒ option (i).
DS
Dr. Saurabh Joshi
PhD Enzymology, NCL Pune
Verified Expert
Verifying A. Enzymes routinely speed up reactions by
factors of 106 to 1017. At body temperature, sucrose is
hydrolysed by sucrase in a few seconds, whereas spontaneous
acid-catalysed hydrolysis would take hours to days. The
effective attack of substrates by reagents (water, O2,
NAD+, etc.) in the presence of enzymes is therefore an
experimentally well-established fact. A is true.
Verifying R. Each enzyme has a small pocket (the
active site) whose three-dimensional shape and chemistry are
complementary to its substrate. When the substrate diffuses
into this pocket, a network of H-bonds, salt bridges and
hydrophobic contacts holds it in a precise position. In the
``induced-fit'' refinement, the enzyme also adjusts its shape
slightly to embrace the substrate more tightly. So the active
site genuinely positions the substrate. R is true.
Why R explains A. The reason an enzymatic reaction is
so fast is exactly the precise positioning provided by the
active site. With the substrate held in a transition-state-like
geometry, only a tiny energy push is needed to convert it to
product; catalytic residues or co-factors sit at the perfect
distance and angle to deliver that push. In other words, R is
the mechanistic cause of A, not just a correlated fact.
Therefore the correct A-R choice is option (i): both correct
and R explains A.
Active-site positioning is the mechanistic cause of effective enzyme catalysis ⇒ both A and R correct, R explains A ⇒ option (i).
VI. Long Answer Type
Q 10.67
Carbohydrates are essential for life in both plants and animals. Name the carbohydrates that are used as storage molecules in plants and animals, also name the carbohydrate which is present in wood or in the fibre of cotton cloth.
Concept used. Carbohydrates serve two great
biological roles: energy storage and structural
support. Plants and animals use chemically different storage
polymers, but both are built from α-D-glucose. Structural
carbohydrates use β-glucose to give rigid, fibrous polymers.
Storage in plants:starch –- a mixture
of linear amylose (α-1,4) and branched
amylopectin (α-1,4 plus α-1,6).
Deposited in seeds, tubers, roots.
Storage in animals:glycogen –-
α-D-glucose units with α-1,4 chain and
very frequent α-1,6 branches, stored in liver and
muscle. Mobilised rapidly by glycogen phosphorylase to
feed blood glucose.
Structural carbohydrate in wood and cotton fibre:cellulose –- linear β-D-glucose polymer
joined by β-1,4 linkages. Adjacent chains H-bond into
microfibrils, giving wood its rigidity and cotton its
tensile strength.
Cell-biology perspective. Carbohydrates serve two
distinct biological purposes: stockpiling chemical energy (storage)
and building load-bearing structures (structural support). The
chemistry that distinguishes the two is the
stereochemistry of the glycosidic bond: α-linkages
give bendable, easily-mobilised polymers; β-linkages give
flat ribbons that aggregate into rigid microfibrils.
[leftmargin=*,nosep]
Plant storage:starch –- a mixture
of amylose (∼ 20%, linear α-1,4) and
amylopectin (∼ 80%, branched α-1,4 plus
α-1,6). Stored in starch grains inside seeds,
tubers (potato), roots (cassava) and rice/wheat
endosperm. Hydrolysed by amylases to release glucose
when the plant needs energy.
Animal storage:glycogen –-
same α-1,4 chain +α-1,6 branches but
with branch points every ∼ 10 residues (denser
branching than amylopectin). Major depots are
liver (∼ 100 g) and skeletal muscle
(∼ 400 g in a 70-kg adult). Glycogen
phosphorylase mobilises glucose-1-phosphate on demand.
Structural in wood and cotton:cellulose
–- a linear β-1,4 polymer of D-glucose. Adjacent
chains H-bond into microfibrils, which aggregate into
macrofibrils, then into the plant cell wall. Wood is
40--50% cellulose; cotton fibre is over 90%
pure cellulose. The straight, ribbon-like geometry of the
β-1,4 chain is what gives cotton its tensile
strength and wood its rigidity.
Why α for storage, β for structure? The
α-1,4 bond bends the chain into a helical coil, allowing
the polymer to pack into compact, easily mobilised granules.
The β-1,4 bond keeps the chain flat and extended; many
such chains can stack tightly through H-bonds into water-resistant
fibres. Evolution chose α for energy reserves and β
for cell-wall reinforcement.
Explain the terms primary and secondary structure of proteins. What is the difference between α-helix and β-pleated sheet structure of proteins?
Concept used. Proteins are organised in a hierarchy.
The primary structure is the specific linear
sequence of amino-acid residues joined by peptide
(-CO-NH-) bonds. The secondary structure is the
regular local folding pattern of the polypeptide backbone, driven
by hydrogen bonds between N-H and C=O
groups. Two main secondary patterns occur: α-helix and
β-pleated sheet.
Primary: sequence of amino acids; held by covalent
peptide bonds. Defines the protein uniquely.
Secondary: regular geometry of the backbone;
held by intra/inter-chain H-bonds.
α-Helix: a right-handed coil; H-bonds
run within a single chain between residue i's
C=O and residue (i+4)'s N-H; side chains
project outward; example: keratin in hair.
β-Pleated sheet: polypeptide chains lie
side-by-side (parallel or antiparallel); H-bonds
run between adjacent chains; the backbone zig-zags
into a pleated sheet; example: silk fibroin.
Hierarchical view. Proteins are organised in four levels.
Primary structure is purely about covalent
connectivity –- the exact order of the 20 standard amino acids
joined head-to-tail by peptide (-CO-NH-) bonds. Mutate even
one residue and you get a different primary structure –- the
substitution of valine for glutamate at position 6 of β-
haemoglobin causes sickle-cell anaemia. Secondary
structure, in contrast, is about non-covalent backbone
folding into regular geometric patterns, stabilised by
N-H ⋯ O=C hydrogen bonds.
α-Helix in detail. The α-helix is a
right-handed coil with 3.6 residues per turn and an axial
translation of 1.5 per residue (helix rise
5.4 per turn). Every C=O of residue i
H-bonds to the N-H of residue i+4, so H-bonds run
within a single chain and parallel to the helix axis.
Side chains project outward. Example: α-keratin in hair,
nails and feathers; myosin in muscle.
β-Pleated sheet in detail. Polypeptide chains
extend nearly fully (translation 3.5 per
residue) and lie side by side, either parallel (same N→C
direction) or antiparallel (opposite directions). H-bonds run
between adjacent strands, perpendicular to the strand
direction. The backbone zig-zags up-and-down, giving the
pleated appearance. Side chains alternate above and below the
sheet. Example: silk fibroin in spider silk and cocoon thread.
Bottom-line difference.α-helix = a single
chain coiled; H-bonds inside. β-sheet = many chains
flat side-by-side; H-bonds between. The choice of pattern is
dictated by the amino-acid sequence: Pro/Gly disrupt
α-helices; small side chains (Gly, Ala, Ser) favour
β-sheets. Both patterns combine in real proteins to give
the unique 3-D tertiary fold.
Write the reactions of D-glucose which can't be explained by its open-chain structure. How can the cyclic structure of glucose explain these reactions?
Concept used. Some experimental observations on glucose
cannot be explained by the simple Fischer (open-chain
-CHO) structure. They become natural once glucose is
represented in the cyclic pyranose hemiacetal form
where C-1 carries a ring oxygen and a hemiacetal -OH.
Failure to give Schiff's test (and weak 2,4-DNP
test): the open-chain structure predicts a free
-CHO that should react with fuchsin – but glucose
doesn't do so reliably. The cyclic form explains this:
the -CHO exists in ∼ 0.02% at equilibrium,
too low for a clear positive Schiff's reaction.
Mutarotation: when crystalline α-D-glucose
([α]D = +112∘) is dissolved in water, the
specific rotation slowly drifts to +52.5∘;
crystalline β (+19∘) drifts to the same
+52.5∘. The open-chain Fischer form cannot
explain this. Cyclic form explains it: the two
anomers interconvert via the open chain until they
equilibrate.
Two crystalline forms of glucose: pure
α- and β-D-glucose crystallise as distinct
solids with different m.p.s and [α]D. Open chain
would predict only one form. Cyclic form explains it:
two anomeric stereoisomers at C-1.
Pentaacetate fails the oxime / Schiff's test:
the open chain would predict that acetylating the five
-OHs would leave the -CHO untouched and
still able to give an oxime. Experimentally, pentaacetate
does not give an oxime. Cyclic form explains it:
once the C-1 hemiacetal -OH is acetylated, C-1
becomes a full acetal and cannot re-open to -CHO.
Mutarotation, two anomeric crystalline forms, pentaacetate's failure to give an oxime, and the weak Schiff's test all need the cyclic hemiacetal structure.
AM
Aishwarya Menon
M.Sc Biochemistry, IISc Bangalore
Verified Expert
Putting all four observations together.
[leftmargin=*,nosep]
Schiff's test failure. Schiff's reagent
(decolourised fuchsin) gives a magenta colour with free
-CHO at high concentration. Glucose at ∼ 99.98%
cyclic form has too little open-chain aldehyde to drive
the reaction. Open-chain Fischer would predict a strong
positive test –- contradicted by observation.
Mutarotation. Pure crystalline α-D-
glucopyranose has [α]D = +112;
pure β has +19. In water both
equilibrate at +52.5. The intermediate
is the open-chain aldehyde, but the two stable
starting solids are cyclic anomers. The Fischer open-
chain structure offers no explanation; the cyclic
hemiacetal explains both forms and their interconversion.
Two crystalline forms. The existence of
α- and β-D-glucose as distinct, isolable
crystalline solids with different melting points
(146 vs 150)
and different specific rotations demands two distinct
stereoisomers at C-1. Only a cyclic structure provides
the new stereocentre.
Pentaacetate inert to NH2OH / Schiff.
After acetylating glucose with (CH3CO)2O, the
product fails the oxime and Schiff's tests. Open-chain
Fischer cannot explain this –- the -CHO should
survive. Cyclic form explains it: the C-1 hemiacetal
-OH has been acetylated, locking C-1 as a full
acetal that cannot revert to -CHO.
Cyclic-structure proposal. The C-5 -OH attacks
the C-1 aldehyde to form a 6-membered pyranose hemiacetal. The
new stereocentre at C-1 has two possible orientations: α
(OH below the ring plane in Haworth projection) and β (OH
above). These two anomers explain mutarotation, the two
crystalline forms, and the pentaacetate's failure to give an
oxime.
Cyclic pyranose hemiacetal explains: weak Schiff's test, mutarotation, two crystalline anomers, and pentaacetate's failure to give oxime. Open-chain Fischer cannot.
Q 10.70
On the basis of which evidences was D-glucose assigned its open-chain structure CH2OH-(CHOH)4-CHO?
Concept used. The classical proof of the open-chain
structure of D-glucose rests on five experimental
evidences: (i) molecular formula; (ii) the HI-reduction
test; (iii) the bromine-water oxidation test; (iv) the
conc. HNO3 oxidation test; (v) reactions with carbonyl
reagents and acetic anhydride.
Molecular formula. Quantitative analysis fixes
glucose as C6H12O6⇒ six carbons, six
oxygens.
HI reduction →n-hexane. Prolonged
HI/Δ replaces every -OH with -H
and reduces -CHO → -CH3; product is
n-hexane ⇒ all 6 C in a single straight chain.
Br2/H2O oxidation → gluconic acid.
Mild oxidation converts -CHO to -COOH⇒ carbonyl at C-1 is an aldehyde.
Conc. HNO3 oxidation → saccharic
acid (dicarboxylic). Both terminal C are oxidised, so
the molecule has a -CH2OH at C-6 (primary -OH)
as well as a -CHO at C-1.
Acetylation → pentaacetate. With acetic
anhydride, exactly five acetate esters form
⇒ five -OH groups in glucose.
Reactions with NH2OH and HCN:
glucose forms a monoxime and a cyanohydrin
⇒ one reactive C=O group, an aldehyde.
Open-chain structure:CH2OH-(CHOH)4-CHO.
Open-chain glucose = 6 C straight chain + -CHO at C-1 + -CH2OH at C-6 + 4 secondary -OHs.
DV
Dr. Vikram Saini
PhD Organic Chemistry, IISc Bangalore
Verified Expert
Building the structure piece by piece.
Carbon skeleton. Quantitative combustion and freezing-
point-depression molar-mass measurements give C6H12O6.
Heating with conc. HI gives n-hexane
CH3-(CH2)4-CH3 –- proving the six carbons are connected
in a single straight chain, with no branching.
Carbonyl group at C-1. Bromine water (a mild oxidant)
converts glucose to gluconic acid, a 6-carbon monocarboxylic
acid HOCH2-(CHOH)4-COOH. Aldehydes oxidise easily to
-COOH under these conditions but ketones do not, so the
carbonyl must be an aldehyde, at one end of the chain (C-1).
Confirmed by formation of a monoxime with NH2OH and a
cyanohydrin with HCN (both diagnostic for one reactive
C=O).
Primary alcohol at C-6. Concentrated HNO3
oxidises both terminal -CHOand the terminal
-CH2OH to -COOH, giving a dicarboxylic
saccharic acid HOOC-(CHOH)4-COOH. The second
-COOH can only come from a primary alcohol at the
opposite end of the chain (C-6). The four middle C's, which
remain -CHOH, must be secondary alcohols.
Five hydroxyl groups. Treatment with acetic anhydride
gives a pentaacetate. Five acetate groups means five free
-OHs on glucose: one primary (C-6) and four secondary
(C-2 to C-5).
Putting it together. 6 C in a straight chain, one
-CHO at C-1, four -CHOHs at C-2 to C-5, one
-CH2OH at C-6 ⇒ the open-chain structure
CHO-CHOH-CHOH-CHOH-CHOH-CH2OH
≡
CH2OH-(CHOH)4-CHO.
The stereochemistry at each chiral C is settled by additional
experiments (Fischer's ingenious sugar work in 1891 using
Killiani–Fischer ascent and Ruff degradation), but the
constitutional structure is established by the five
evidences listed.
Glucose open-chain structure CH2OH-(CHOH)4-CHO is fixed by molecular formula, HI reduction to n-hexane, Br2/H2O to gluconic acid, conc. HNO3 to saccharic acid, pentaacetate formation, and oxime/cyanohydrin formation.
Q 10.71
Write the structures of fragments produced on complete hydrolysis of DNA. How are they linked in DNA molecule? Draw a diagram to show pairing of nucleotide bases in the double helix of DNA.
Concept used. Complete hydrolysis of DNA breaks it into
three classes of fragments: nitrogenous bases (A, T, G,
C); 2-deoxyribose (the pentose sugar); and
phosphoric acid (H3PO4). In intact DNA these
fragments are linked by two kinds of bonds:
[label=(),leftmargin=*]
N-glycosidic bond: base attached at C-1'
of the sugar (purines via N9, pyrimidines via N1).
Phosphodiester bond: phosphate bridges the
3'-OH of one sugar to the 5'-OH of the next sugar
⇒ backbone of the chain.
The two strands of DNA pair via Watson–Crick hydrogen
bonds: A=T (2 H-bonds), G≡ C (3 H-bonds).
Hydrolysis fragments: base + 2-deoxyribose + H3PO4.
Inside DNA: base–sugar via N-glycosidic bond
⇒ nucleoside.
Hydrolysis products. Complete acid hydrolysis of DNA
yields three classes of small molecules:
[leftmargin=*,nosep]
Phosphoric acid (H3PO4): from the
phosphate backbone.
2-Deoxyribose (C5H10O4): the pentose
sugar of every DNA nucleotide.
Nitrogenous bases: the two purines
(adenine A, guanine G) and two
pyrimidines (cytosine C, thymine T).
Linkages inside DNA. Inside intact DNA, the three
fragments are joined by two kinds of bonds:
[leftmargin=*,nosep]
Each base is attached to the C-1' of 2-deoxyribose
by an N-glycosidic bond (purines through N-9,
pyrimidines through N-1). The base-sugar unit is called
a nucleoside (deoxyadenosine, deoxyguanosine,
deoxycytidine, thymidine).
Each sugar carries a phosphate group at C-5' ester-bonded
as -O-PO3H-. When this phosphate also esterifies the
3'-OH of the next sugar, a phosphodiester
bond forms, linking nucleotide to nucleotide along
the chain. The full polymer is built up
5'→ 3'.
Watson–Crick base pairing. Two complementary
polynucleotide strands wind around each other to form a
right-handed double helix. The strands are antiparallel:
one runs 5'→ 3', the other 3'→ 5'. The bases project
inward and pair through hydrogen bonds:
[leftmargin=*,nosep]
Adenine ≡ Thymine: 2 hydrogen bonds.
Guanine ≡ Cytosine: 3 hydrogen bonds.
A purine always pairs with a pyrimidine, so every ``rung'' of
the helix is the same width (∼1.08).
This complementary pairing is the chemical basis of genetic
information storage and the templating of DNA replication.
Biomolecules Class 12 Chemistry Exemplar Solutions FAQs
Q. How many problems are there in the Class 12 Chemistry Chapter 10 Biomolecules Exemplar?
The Biomolecules Exemplar has 25 representative problems across MCQ-I (8), MCQ-II (4), Short Answer (8), Matching (1) and Assertion-Reason / LA (4). The Collegedunia PDF works each item with a Solution plus an Expert's Solution.
Q. Is Biomolecules Chapter 10 or Chapter 14 in NCERT?
Under the current 2026-27 NCERT, Biomolecules is Chapter 10 of Class 12 Chemistry. Older prints and many third-party sites still list it as Chapter 14, but the content of the chapter is the same, with the "Hormones" section trimmed in the new edition.
Q. What is the CBSE weightage of Biomolecules in the Class 12 board exam?
The chapter carries roughly 3 to 5 marks, usually as one 2-mark VSA on a named biomolecule, vitamin or deficiency disease, and a 3-mark SA on protein structure, glycosidic linkage or DNA versus RNA in alternate years.
Q. Which topics from Biomolecules are most important for JEE Main and NEET?
The highest-yield topics are carbohydrate classification (mono- / di- / polysaccharides, reducing vs non-reducing), polysaccharide linkages (α-1,4 vs β-1,4, branching by α-1,6), protein primary to quaternary hierarchy and α-helix H-bonding, DNA versus RNA base set and the 5′-3′ phosphodiester linkage, and vitamin classification with deficiency diseases.
Q. Why is sucrose a non-reducing sugar even though it is built from glucose and fructose?
Sucrose joins the anomeric C1 of α-D-glucose to the anomeric C2 of β-D-fructose through an α,β-1,2 glycosidic bond. This bond locks up both anomeric carbons, so neither sugar can open its hemiacetal ring to expose a free -CHO or α-hydroxy ketone group. Without a free anomeric carbon there is nothing for Fehling or Tollens reagent to oxidise, so sucrose is non-reducing. Maltose and lactose retain one free anomeric C and are reducing.
Q. What stabilises the α-helix structure of proteins?
The α-helix is held by intra-chain hydrogen bonds between the N−H of every residue i and the C=O of residue i+4. These H-bonds run parallel to the helix axis and lock the right-handed coil. Peptide bonds form the primary backbone, van der Waals forces are too weak, and disulphide bridges stabilise tertiary structure - not the helix itself.
Q. What is the difference between a nucleoside and a nucleotide?
A nucleoside is a base joined to a sugar (no phosphate). A nucleotide is a base joined to a sugar joined to a phosphate. Nucleic acids (DNA and RNA) are built only from nucleotides because the phosphate is the bridge that condenses with the 3′-OH of the next sugar to form a 5′ to 3′ phosphodiester linkage. Mnemonic: nucleoside = NO-side (no phosphate); nucleotide = tide of phosphate.
Q. Are the Exemplar problems on Biomolecules harder than the NCERT textbook exercises?
Yes. The Exemplar reframes textbook facts as multi-factor classification puzzles, asks for comparison of bonds across protein levels, and tests assertion-reason logic on configuration versus rotation. The Collegedunia Exemplar Solutions PDF works each item with a Solution plus an Expert's Solution that names the controlling rule.
Q. How do I download the Biomolecules Exemplar Solutions PDF for free?
Use the download button at the top of this page to get the free PDF of NCERT Exemplar Solutions for Class 12 Chemistry Chapter 10 Biomolecules, fully aligned to the 2026-27 syllabus.
Q. What is the difference between anomers, epimers and enantiomers in carbohydrate chemistry?
Anomers, epimers, and enantiomers are all types of stereoisomers but they differ in which carbons swap configuration. Anomers differ only at the anomeric carbon (C1 in aldoses, C2 in ketoses) - the new chiral centre created when the open chain cyclises; alpha- and beta-D-glucopyranose are anomers. Epimers differ at one non-anomeric chiral carbon - glucose and galactose are C4 epimers; glucose and mannose are C2 epimers. Enantiomers are non-superimposable mirror images; the chirality flips at every chiral centre (e.g. D-glucose and L-glucose). The Collegedunia Exemplar Solutions PDF flags each of these terms inside the relevant solution.
Q. What is mutarotation and why does freshly dissolved alpha-D-glucose show a changing optical rotation?
Mutarotation is the gradual change in optical rotation of a freshly dissolved pure anomer of a reducing sugar as it equilibrates with the other anomer through the open-chain form. Pure alpha-D-glucopyranose has [α] = +112 degrees; on dissolving in water it equilibrates with beta-D-glucopyranose ([α] = +19 degrees) via the open-chain aldehyde, giving an equilibrium rotation of +52.5 degrees. The equilibrium composition is about 36% alpha, 64% beta, and trace open-chain. Mutarotation only occurs in reducing sugars because non-reducing sugars (like sucrose) cannot open back to the chain form.
Q. How do purines differ from pyrimidines and which bases are unique to DNA vs RNA?
Purines are double-ring nitrogen bases - adenine (A) and guanine (G); mnemonic "PURe As Gold". Pyrimidines are single-ring nitrogen bases - cytosine (C), thymine (T) and uracil (U). DNA contains A, G, C, T (thymine is found only in DNA); RNA contains A, G, C, U (uracil is found only in RNA). Cytosine is in both. In the double helix, Watson-Crick pairs are A ··· T via 2 H-bonds and G ··· C via 3 H-bonds; each pair is one purine + one pyrimidine, so the pair widths match across the helix.
Q. Which vitamins are fat-soluble vs water-soluble and what deficiency disease does each cause?
The four fat-soluble (ADEK) vitamins are A, D, E, K, stored in the liver and adipose tissue. The water-soluble vitamins are the B-complex (B1, B2, B6, B12, etc.) and Vitamin C, excreted in urine and supplied daily (exception: B12 is stored). Disease-vitamin pairs that the Exemplar most-frequently tests: A - night blindness / xerophthalmia; B1 (thiamine) - beri-beri; B2 - cheilosis; B6 - anaemia / convulsions; B12 (cobalamin) - pernicious anaemia; C (ascorbic acid) - scurvy; D - rickets / osteomalacia; E - sterility, muscular weakness; K - poor blood clotting.
Q. Why is haemoglobin chosen as the textbook example of quaternary protein structure?
Haemoglobin is a tetramer built from two alpha chains (141 residues each) and two beta chains (146 residues each); each subunit cradles a heme group with an Fe(II) centre. The four subunits associate through non-covalent contacts and a few salt bridges to give a defined 3-D assembly with molecular mass about 64,500 u. Binding of one O2 molecule increases the affinity for the next - the classic positive cooperativity that gives haemoglobin a sigmoidal O2-binding curve, unlike monomeric myoglobin. The multi-chain assembly with cooperative function makes haemoglobin the canonical NCERT example of quaternary structure.

































































































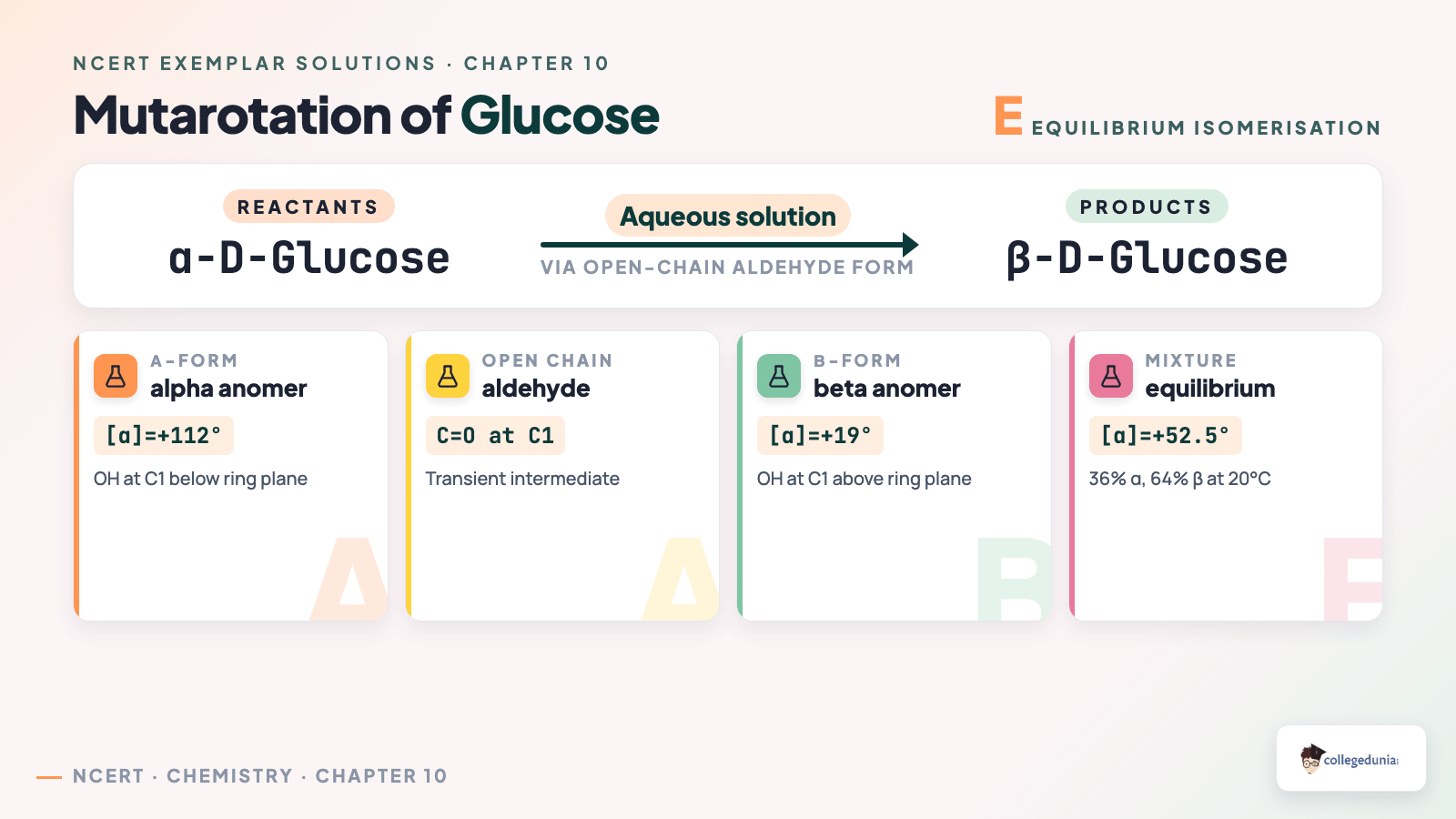



Comments