Biology Mentor, Hindu College | Updated on - Jul 21, 2026
Biotechnology: Principles and Processes turns recombinant DNA and bioreactors into real products like insulin. Class 12 Biology Chapter 9 covers the full toolkit, from restriction enzymes to gel electrophoresis. This page hosts the step-by-step NCERT Solutions PDF.
Why Biotechnology Principles and Processes is a Score-Boost Chapter for NEET 2027
This is a process-based chapter that maps directly onto NEET MCQ stems. With named restriction enzymes, cloning vectors and a fixed rDNA workflow, the patterns repeat year on year.
NEET pulled 4 direct-recall MCQs in 2025 and 5 in 2024, mostly on enzyme nomenclature and gel electrophoresis.
Biotechnology Principles and Processes NCERT Solutions: Exercise Breakdown
The 11 questions sit in one exercise. The table maps them across the four NCERT sub-topics by NEET frequency.
Sub-Topic (NCERT section)
NCERT Q Numbers
Question Count
NEET Yield (last 5 yrs)
Principles of Biotechnology (9.1)
Q1, Q2
2
2-3 questions
Tools of rDNA technology: enzymes, vectors, host (9.2)
Q3, Q4, Q5, Q6
4
6-8 questions
Processes: cutting, amplification, ligation, transformation, gel run (9.3)
Q7, Q8, Q10
3
4-5 questions
Bioreactors and downstream processing (9.3 cont.)
Q9, Q11
2
1-2 questions
Tools of rDNA technology (9.2) is the highest-yield NEET sub-topic, so prioritise Q3, Q5 and Q6. Q9 on bioreactors is a near-guaranteed NEET item.
Biotechnology Principles and Processes Class 12 Biology PYQ Trend (2025 to 2021)
The breakdown below maps this chapter across CBSE Boards and NEET over five cycles.
Year
CBSE Class 12 Boards
NEET
Most-Asked Topic
2025
6 marks (pBR322 + PCR)
4 questions
EcoRI / pBR322
2024
7 marks (rDNA workflow + bioreactor)
5 questions
Restriction enzyme / Taq
2023
5 marks
3 questions
Sticky ends / DNA ligase
2022
6 marks
4 questions
Gel electrophoresis / vector
2021
5 marks
3 questions
Ti plasmid / transformation
The five-year average is 5.8 marks in CBSE and 3.8 questions in NEET, so prepare Q3 to Q8 first.
NEET prep tip: Names like EcoRI, HindIII, Agrobacterium tumefaciens and Thermus aquaticus are direct-recall MCQs. In EcoRI the Roman "I" and capital "R" are part of the nomenclature; one wrong letter loses the mark.
Sample Fully-Solved Question: Restriction Enzymes and Palindromes (Q5)
The NCERT question asks: "Do eukaryotic cells have restriction endonucleases? Justify your answer." The four-mark CBSE pattern is below.
Step 1 (1 mark). A restriction endonuclease cuts the sugar-phosphate backbone at a specific palindromic site.
Step 2 (1 mark). Eukaryotic cells do not have them. Their DNA is methylated and sits inside a nuclear envelope, safe from viral DNA.
Step 3 (1 mark). In bacteria they form a restriction-modification (R-M) system that degrades invading viral DNA.
Step 4 (1 mark). Biologists use them in vitro to cut the gene and vector at the same palindrome, making sticky ends that DNA ligase joins. EcoRI cuts at 5'-G/AATTC-3'.
CBSE 2024 awarded zero marks to scripts that wrote "eukaryotes do not need them" without naming methylation or the nuclear envelope. The mechanism word is mandatory.
Where Students Lose Marks in Biotechnology Principles and Processes (Class 12 Biology)
Students remember the names but mis-state the cut site. The mistakes below cost the most marks.
Mistake 1. Writing "EcoR1" with digit 1 instead of Roman "I". The Roman numeral is mandatory.
Mistake 2. Confusing DNA ligase (joins phosphodiester bonds) with DNA polymerase (synthesises a new strand).
Mistake 3. Calling pBR322 a virus. It is a plasmid, a circular bacterial DNA used as a cloning vector.
Mistake 4. Listing only "PCR" without the three steps: denaturation, annealing and extension (~72 degree C with Taq polymerase).
Mistake 5. Spelling "Thermus aquaticus" wrong. A NEET trap on the source of Taq polymerase.
Top Tools-of-rDNA Recall Table for Class 12 Biology Chapter 9
Memorise the enzyme in italics, the exact recognition site and the source.
How to Study Biotechnology Principles and Processes for Class 12 Biology Boards
Bioreactors and downstream processing are often under-prepared, yet NEET tests both nearly every year. The three-day plan below splits the 11 questions by frequency.
Day
Focus
NCERT Q to Solve
Time
Day 1
Principles + Tools (9.1 to 9.2): enzymes, vectors, host
Q1, Q2, Q3, Q4, Q5, Q6
3 hours
Day 2
Processes of rDNA (9.3): cutting, amplification, ligation, transformation, gel run
Q7, Q8, Q10
3 hours
Day 3
Bioreactors + downstream + full revision + 1 PYP
Q9, Q11
2 hours
That is about 8 hours over 3 days, ending with one NEET-pattern paper.
All NCERT Solutions for Biotechnology Principles and Processes with Step-by-Step Working
Every NCERT textbook question for Class 12 Biology Chapter 9 Biotechnology Principles and Processes is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
NCERT Solutions - Class 12 Biology Chapter 9
Q 9.1
Can you list 10 recombinant proteins which are used in medical practice? Find out where they are used as therapeutics (use the internet).
Concept used. A recombinant protein is any protein
produced by expressing a cloned gene in a heterologous host (typically
E. coli, yeast, or mammalian cell lines). Because the gene is
inserted into a host through recombinant DNA technology,
the protein it codes for is identical to (or a deliberately modified
version of) the human/animal protein, but can be manufactured in
industrial quantities at controlled purity. Medical recombinant
proteins replace a missing endogenous protein, neutralise a pathogen,
or act as a biological signal.
Why recombinant, not extracted?
Extracting insulin from cattle/pig pancreas works, but the animal
protein differs in 1–3 amino acids and triggers immune reactions in
∼ 5% of patients. Cloning the human insulin gene in
E. coli gives exactly the human sequence with zero animal
contamination.
Ten recombinant therapeutic proteins commonly named
in NCERT-aligned references:
Human Insulin (Humulin) - treats type-1 and
advanced type-2 diabetes mellitus.
Human Growth Hormone (Somatotropin / hGH)
- treats pituitary dwarfism and Turner syndrome.
Erythropoietin (EPO) - treats anaemia in
chronic kidney disease and chemotherapy patients.
Interferon α, β, γ -
antiviral therapy in chronic hepatitis B/C, and
disease-modifying therapy in multiple sclerosis.
Tissue Plasminogen Activator (tPA / Alteplase)
- clot-buster used within hours of an acute
ischaemic stroke or myocardial infarction.
Factor VIII - corrects clotting deficiency
in haemophilia A.
Factor IX - corrects clotting deficiency in
haemophilia B (Christmas disease).
Hepatitis B surface antigen (HBsAg) vaccine
- prevents hepatitis B infection; the first widely
used recombinant vaccine.
Monoclonal antibodies such as
Trastuzumab (HER2-positive breast cancer)
and Rituximab (non-Hodgkin lymphoma,
rheumatoid arthritis).
Why this list matters. Each protein here was first
purified in milligram amounts from human/animal sources at
prohibitive cost. Recombinant production in microbial or
mammalian bioreactors dropped the cost per dose by 10–100×
and removed the risk of carrying through donor-derived
viruses (HIV, hepatitis) that haunted plasma-derived clotting
factors in the 1980s.
Strategic angle (clinical-pharmacology framing). Rather than
listing proteins at random, group them by the deficiency they correct
or the pathway they target. This anchors each name to a clinical
endpoint that examiners reward.
Replacement therapies (give back a missing protein):
Insulin (diabetes), hGH (dwarfism), EPO (anaemia in renal
failure), Factor VIII (haemophilia A), Factor IX (haemophilia
B). Each gene is cloned and expressed because the natural
source is either insufficient or unsafe.
Signalling/immune-modulator therapies: Interferons
(α, β, γ) for chronic viral infections and
multiple sclerosis; G-CSF (Filgrastim) to rescue bone marrow
after chemotherapy.
Acute thrombolytic: Tissue plasminogen activator
(Alteplase) dissolves fibrin clots in stroke and myocardial
infarction. Cloned from human cDNA and expressed in CHO
cells (Chinese-Hamster-Ovary mammalian line) because the
glycosylation pattern matters for activity.
Vaccines and antibodies: HBsAg subunit vaccine
(Saccharomyces cerevisiae host) was the first
recombinant vaccine; monoclonals such as Trastuzumab (anti-HER2)
and Rituximab (anti-CD20) target tumour-cell surface markers.
Why the host matters. Bacteria are cheap but cannot
glycosylate; yeast can; mammalian cells (CHO, HEK293) give
human-like post-translational modifications and are chosen
whenever the protein is folded, glycosylated or
disulphide-rich (antibodies, EPO, Factor VIII).
Why this matters. The category-wise grouping mirrors how
pharmacology textbooks classify these molecules and the host-choice
logic is a frequent NEET-PG / GATE Biotechnology question.
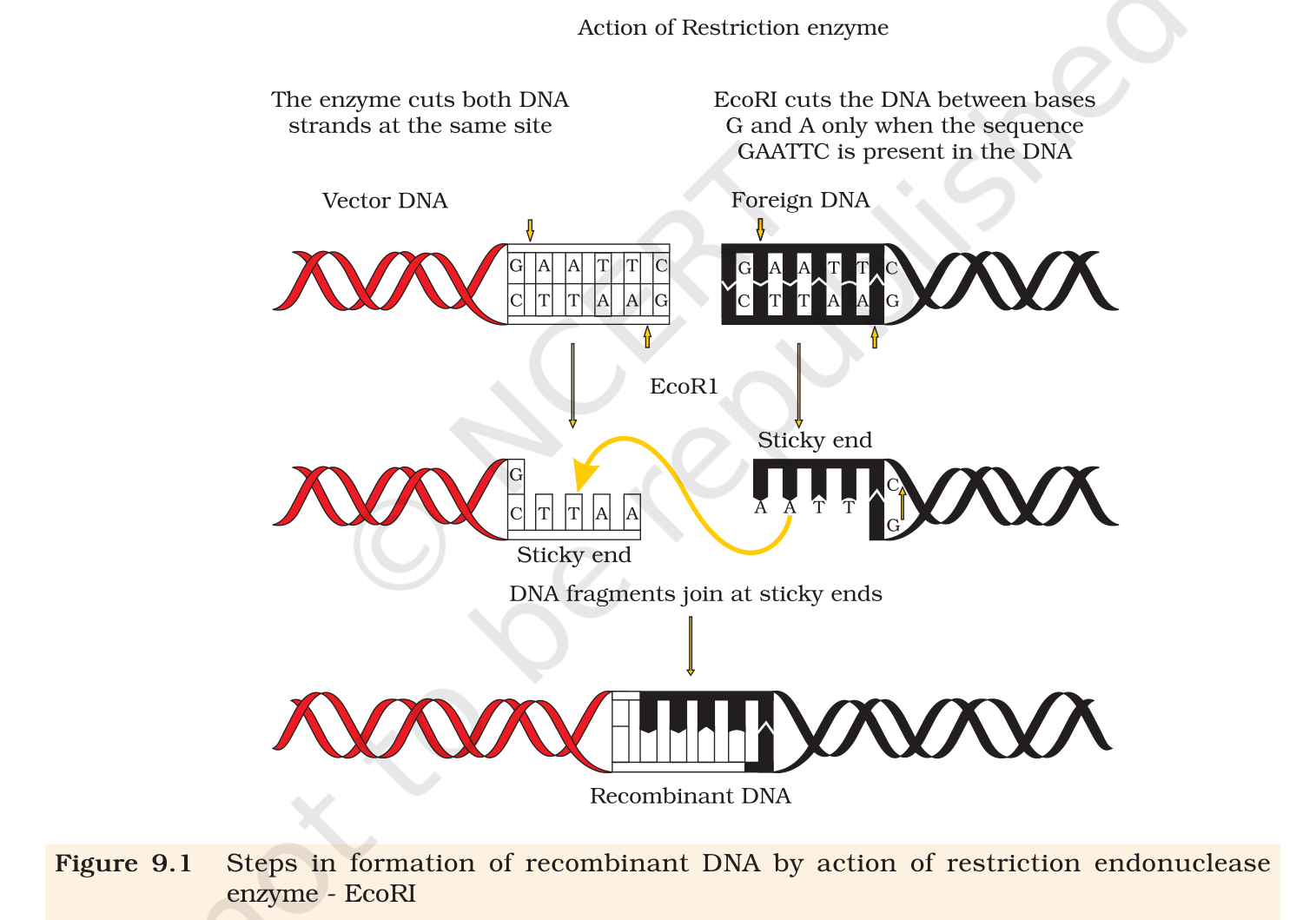
Make a chart (with diagrammatic representation) showing a restriction enzyme, the substrate DNA on which it acts, the site at which it cuts DNA and the product it produces.
Concept used. A restriction endonuclease is a
bacterial enzyme that scans double-stranded DNA, recognises a specific
short palindromic sequence (usually 4–8 base pairs), and
cleaves the sugar–phosphate backbone of both strands inside or
adjacent to that recognition site. The most studied member is
EcoRI from Escherichia coli, which recognises the
hexamer 5'-GAATTC-3' and cuts between G and A on each strand,
leaving 4-nucleotide single-stranded overhangs called
sticky ends. Two DNA fragments cut by the same enzyme have
complementary sticky ends and can therefore be joined by DNA ligase.
Substrate. Any double-stranded DNA molecule
containing the hexanucleotide recognition site. Example below:
a plasmid (vector) and a foreign DNA fragment, both
containing one 5'-GAATTC-3' site.
Recognition site (palindrome). Reading 5'→ 3'
on either strand gives the same sequence:
arrayc
5' GAATTC 3'
3' CTTAAG 5'
array
Site of cleavage. EcoRI cuts the phosphodiester
bond between G and A on each strand, indicated by
the arrows below:
arrayc
5' GATTC 3'
3' CTTAA5'
array
Because the cuts are staggered (not directly opposite each
other), each fragment carries a four-base single-stranded
5'-overhang.
Products. Two DNA fragments with complementary
5'-AATT-3' sticky ends:
5' G AATT C 3'
3' CTTAA G 5'
Mixing vector-cut DNA with foreign-DNA-cut fragments allows
their AATT overhangs to base-pair; DNA ligase then
seals the nicks to form a single recombinant DNA
molecule.
Fig. 9.1, NCERT Class 12 Biology, Chapter 9. Action of restriction enzyme EcoRI producing sticky ends and the resulting recombinant DNA.
[See diagram in the PDF version]
EcoRI recognises 5'-GAATTC-3', cuts between G and A on
both strands, and produces DNA fragments with 4-base 5'-AATT
sticky ends that ligate to give recombinant DNA.
PR
Priya Reddy
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle (chart-format framing). Examiners love a
clean two-row table: enzyme name + source on the left, substrate /
cut-site / product on the right. Present the four required pieces of
information explicitly.
Enzyme + source.EcoRI, isolated from
Escherichia coli strain RY13. Naming convention: first
letter of genus (E), first two of species (co), strain (R),
order of discovery (I).
Substrate. Double-stranded DNA bearing the
palindromic recognition hexamer GAATTC; could be a viral
genome, plasmid or any genomic DNA.
Cut site. Between the 5'-G and the next A on each
strand. Because the cuts on the two strands are offset by
four bases, the enzyme makes a staggered cut rather
than a flush cut.
Product. Two DNA fragments, each ending in a
5'-AATT single-stranded overhang. These complementary
overhangs are sticky ends - they base-pair
spontaneously with any other fragment cut by EcoRI, allowing
recombination across species barriers.
Why sticky ends matter. They are the reason
recombinant DNA technology works: a human gene cut with
EcoRI and a bacterial plasmid cut with EcoRI will join up
because both carry the same AATT overhang.
Why this matters. The chart should always have four columns:
Enzyme | Source | Recognition + cut site | Product.
Examiners reward the source organism and the staggered-cut detail.
Chart: EcoRI (from E. coli) recognises
5'-G↓AATTC-3' on ds-DNA and produces two fragments
with complementary 5'-AATT sticky ends.
Q 9.3
From what you have learnt, can you tell whether enzymes are bigger or DNA is bigger in molecular size? How did you know?
Concept used.Molecular size can be compared two
ways: by molecular weight (number of atoms / mass in
daltons) or by physical length of the molecule. For
biological macromolecules a genome's DNA is typically thousands to
billions of base-pairs long, while a single enzyme is a folded
polypeptide of a few hundred amino acids. Gel-electrophoresis
behaviour (Section 9.3.1 of the chapter) is the direct evidence: in
an agarose gel the DNA migrates more slowly and gets caught higher
up, while a protein of comparable nominal mass slips through.
Reference numbers
The E. coli genome is ∼ 4.6 × 106 base pairs
⇒ molecular weight ∼ 3 × 109 Da. A typical
restriction-enzyme polypeptide is ∼ 300 amino acids
⇒ molecular weight ∼ 3 × 104 Da
(∼ 30 kDa). Therefore the genomic DNA is roughly 105
times heavier than the enzyme that cuts it.
Compare on the same gel. In agarose-gel
electrophoresis, DNA fragments and proteins behave very
differently. DNA, being uniformly negatively charged and
much larger, migrates only short distances and is held back
in the gel matrix; proteins of a few tens of kDa run far
toward the electrode. The direct visual evidence: DNA stays
near the well, the protein runs away.
Count the monomers. A bacterial chromosome is
∼ 106–107 nucleotide pairs long. Even the
largest enzymes are ≤ 103 amino acids long. By
monomer count alone, DNA wins by 3–4 orders of magnitude.
Translate to mass. Average nucleotide pair ≈
650 Da; average amino acid ≈ 110 Da. So 1 million
bp of DNA ≈ 6.5 × 108 Da, whereas a 300-aa
enzyme ≈ 3.3 × 104 Da. The DNA is
∼ 2 × 104 times heavier than the enzyme.
Visual evidence in the chapter. Section 9.3
figures show one tiny restriction enzyme nibbling at a
much longer DNA strand. The enzyme docks onto a 6-bp
recognition site that occupies a vanishing fraction of the
substrate molecule.
DNA is far bigger than enzymes - by mass (104–105
times) and by physical length (kilobases vs nanometre-scale folded
proteins). The proof: on agarose-gel electrophoresis the DNA stays
near the well while a ∼ 30 kDa enzyme migrates far down.
KB
Karan Banerjee
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Quick-reading angle. The question is testing whether you
remember the gel-electrophoresis intuition and the order-of-magnitude
comparison. Frame the answer as: evidence + numbers + visual.
Evidence from gel electrophoresis. On a 1% agarose
gel run with the same voltage and the same dye, DNA bands
appear near the wells (large molecules can barely thread
through the pore network) while protein bands run off the
bottom unless the gel is much denser (which is why proteins
are usually resolved on polyacrylamide, not agarose).
Numerical comparison. Molecular weight of one
nucleotide pair ≈ 650 Da; molecular weight of one
amino-acid residue ≈ 110 Da. The shortest functional
DNA (a gene of ∼ 1000 bp) already weighs 6.5 ×
105 Da while an enzyme of ∼ 300 residues weighs
only 3.3 × 104 Da. DNA wins by ∼ 20× for
a single gene and by millions of times for a chromosome.
Physical-length comparison. 1 bp of B-form DNA
= 0.34 nm, so a 1-kb gene stretches ∼ 340 nm. A
folded globular enzyme of 300 aa is ∼ 5–8 nm across.
Even unfolded as a polypeptide chain, 300 × 0.38 nm
≈ 114 nm, still well under a single kilobase.
Visualisation. Picture the restriction enzyme as a
small molecular pacman (∼ 30 kDa) docking onto
a substrate DNA strand that is thousands of base-pairs long
- a tiny machine reading along a very long tape.
Why this matters. The size disparity is the reason a single
enzyme can cleave a whole genome at every occurrence of its
6-bp site, generating thousands of fragments from one continuous
piece of DNA.
DNA is much bigger than enzymes; the evidence is the
slow agarose-gel migration of DNA versus the fast migration of any
typical 30-kDa protein.
Q 9.4
What would be the molar concentration of human DNA in a human cell? Consult your teacher.
Concept used.Molar concentration (molarity, M)
is defined as the number of moles of a substance per litre of
solution:
M = nV = NNA · V,
where N is the number of molecules of the substance, NA
≈ 6.022 × 1023 mol-1 is Avogadro's
number, and V is the volume of solution in litres. A diploid
human somatic cell contains two copies of the haploid genome
(one set of 23 chromosomes from each parent), so N = 2 molecules
of nuclear DNA per cell. The remaining unknown is the cell volume,
which for a typical mammalian cell is ∼ 1000 3= 1000 × 10-15 L = 10-12 L.
A Fermi-style estimation
We are estimating an extremely small concentration. Order-of-magnitude
errors in cell volume are fine; the key insight is that with only 2
copies of the genome in a picolitre, the molarity has to be in the
femto-molar range.
Count the DNA molecules per cell. A diploid human
cell contains 46 chromosomes = 2 complete genomes (one
maternal, one paternal). Treating each genome as one DNA
molecule (it is actually 23 linear molecules, but the
question asks for ``human DNA'' as a whole):
N = 2 copies.
Convert to moles. Number of moles of DNA per cell:
n = NNA = 26.022 × 1023
= 3.32 × 10-24 mol.
Estimate cell volume. A typical mammalian cell has
a diameter ∼ 10–20 . Take
V ≈ 1000 3:
V = 1000 × (10-6m)3 = 10-15 m3
= 10-12L.
(Recall 1 m3 = 1000 L.)
Apply the molarity definition.M = nV = 3.32 × 10-24 mol
10-12L
= 3.32 × 10-12 mol/L.
In SI prefixes, M ≈ 3.3 pmol/L, i.e.
∼ 3 picomolar.
Molar concentration of human DNA in a single diploid cell
≈ 3.32 × 10-12M (∼ 3 picomolar).
AN
Aanya Nair
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Strategic angle (Fermi-estimation framing). This is a
back-of-envelope question. The answer hangs on three pieces of
information: number of DNA molecules per cell, Avogadro's number, and
a sensible cell volume. Show each clearly.
Number of DNA molecules. A diploid human somatic
cell carries two complete genomes:
N = 2 molecules of genomic DNA.
(One could refine this to 46 chromosomes; the molarity
scales linearly and only changes the prefactor.)
Convert to moles using Avogadro's number.n = NNA = 26.022 × 1023 mol-1.
Numerator = 2; denominator = 6.022 × 1023;
ratio = 0.332 × 10-23 mol = 3.32 ×
10-24 mol.
Estimate cell volume. Typical somatic-cell diameter
d ≈ 10–15 . Treating the cell as a
sphere of d = 12 :
V = 43π(d2)3
= 43π (6 )3
≈ 9 × 1023 ≈ 10-12L.
Sanity check. The answer is far below the molarity
of typical buffer salts (10-3 M) or even rare metabolites
(10-9 M), which makes sense - there are only two copies
of the genome in a tiny picolitre volume. The relevant
number of molecules, not concentration, is what
matters in molecular biology.
Why this matters. This exercise shows why analytical
techniques (PCR, qPCR) had to be invented: with only ∼ 10-12 M
DNA per cell, you cannot detect a target gene by classical chemistry.
Amplification is mandatory.
Mhuman DNA per cell ≈ 3.3 × 10-12M
(∼ 3 picomolar).
Q 9.5
Do eukaryotic cells have restriction endonucleases? Justify your answer.
Concept used. A restriction endonuclease is a
sequence-specific enzyme that cleaves foreign double-stranded
DNA at a palindromic recognition site, while the host's own
DNA is protected by methylation at the same sites. This pair of
activities (restriction + modification) constitutes a
prokaryotic defence system against invading viruses (bacteriophages):
phage DNA is unmethylated, the host enzymes shred it; the host's own
chromosomal DNA carries methyl groups on key bases (typically the
adenine of GATC or the cytosine of CCGG) and therefore escapes
cleavage. Eukaryotic cells use a fundamentally different anti-viral
defence (RNA interference, type-I interferon signalling, restriction
factors of the APOBEC/TRIM family) and do not encode the
classical sequence-specific restriction enzymes used in genetic
engineering.
Direct answer.No. Eukaryotic cells do
not possess restriction endonucleases of the type
used in recombinant DNA technology (Types I, II, III).
Justification - why prokaryotes have them.
Restriction-modification systems evolved as a microbial
immune system against bacteriophages. Bacteria carry
a methylase and a restriction enzyme that share the same
recognition site: the methylase tags host DNA so it is
ignored; the restriction enzyme cleaves any unmethylated
invading phage DNA into fragments.
Justification - why eukaryotes do not need them.
Eukaryotic cells face viruses but defend through different
mechanisms: RNA interference (siRNA / miRNA pathway
targeting viral mRNA), interferon-induced antiviral state,
cell-autonomous restriction factors (APOBEC3, SAMHD1, TRIM5),
and the adaptive immune system in metazoans. Carrying
promiscuous DNA-cutting enzymes in the cytoplasm would risk
fragmenting their own enormous nuclear genome.
Caveat - eukaryotic nucleases that cut DNA.
Eukaryotes do encode other nucleases: endonuclease G
(apoptotic DNA fragmentation), DNase I/II (extracellular
digestion), Cas9 (in archaea; introduced into
eukaryotes for genome editing). These are not
sequence-specific restriction enzymes - they cut after a
defined biological trigger (apoptosis, RNA guidance), not at
a palindrome.
No. Eukaryotic cells lack the bacterial
restriction-modification systems used in cloning. The enzyme evolved
as a phage-defence tool in prokaryotes; eukaryotes use RNA
interference and the immune system instead.
VS
Vivaan Sharma
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Structural-defence angle. Treat this as a comparative
immunology question: which lineage uses which weapon against
viruses?
Bacteria: restriction–modification systems. A
methylase protects self DNA; an endonuclease cuts unmethylated
invading DNA. The two activities share the same recognition
site (e.g. GATC).
Archaea and bacteria also have CRISPR–Cas: a
sequence-programmable adaptive immune system. Cas9 is the
eukaryote-friendly cousin that we now exploit for genome
editing.
Eukaryotes: do not encode classical restriction
endonucleases. Their genome is huge (3 × 109 bp in
humans) - releasing a promiscuous palindrome-cutter would
shred it. Instead, they encode:
Interferon-stimulated genes (PKR, OAS, IFIT)
inducing an antiviral state.
Restriction factors (APOBEC3G, SAMHD1,
TRIM5α) blocking specific viral life-cycle
steps.
Adaptive immunity in vertebrates (T cells,
B cells, antibodies).
Why the difference? Genome size and architecture.
A 4-Mb bacterial chromosome can be protected by methylating
a few thousand recognition sites; a 3-Gb mammalian genome
cannot tolerate the metabolic burden of methylating millions
of sites just to keep a self-destruct enzyme in check.
Why this matters. The answer also explains why bacterial
hosts (E. coli) must be re-engineered to be restriction-minus
(e.g. DH5α) before they can accept foreign DNA: otherwise
the host's own restriction enzymes would shred the incoming plasmid.
Eukaryotic cells do not have restriction endonucleases
because they use RNA-interference, interferon signalling and
adaptive immunity instead of restriction–modification systems.
Q 9.6
Besides better aeration and mixing properties, what other advantages do stirred tank bioreactors have over shake flasks?
Concept used. A shake flask is a small (50 mL to
2 L) Erlenmeyer-flask culture agitated on an orbital shaker; it
supports research-scale growth but offers no real-time monitoring or
control. A stirred-tank bioreactor is an
industrial-grade vessel (100–1000 L, sometimes much larger)
equipped with an internal stirrer, an aeration sparger, and an
integrated control system that monitors and adjusts pH, temperature,
dissolved oxygen, foam, and substrate feed in real time. Beyond the
obvious aeration/mixing advantage stated in the question, the
control architecture is what makes bioreactors the only viable
platform for industrial-scale recombinant-protein production.
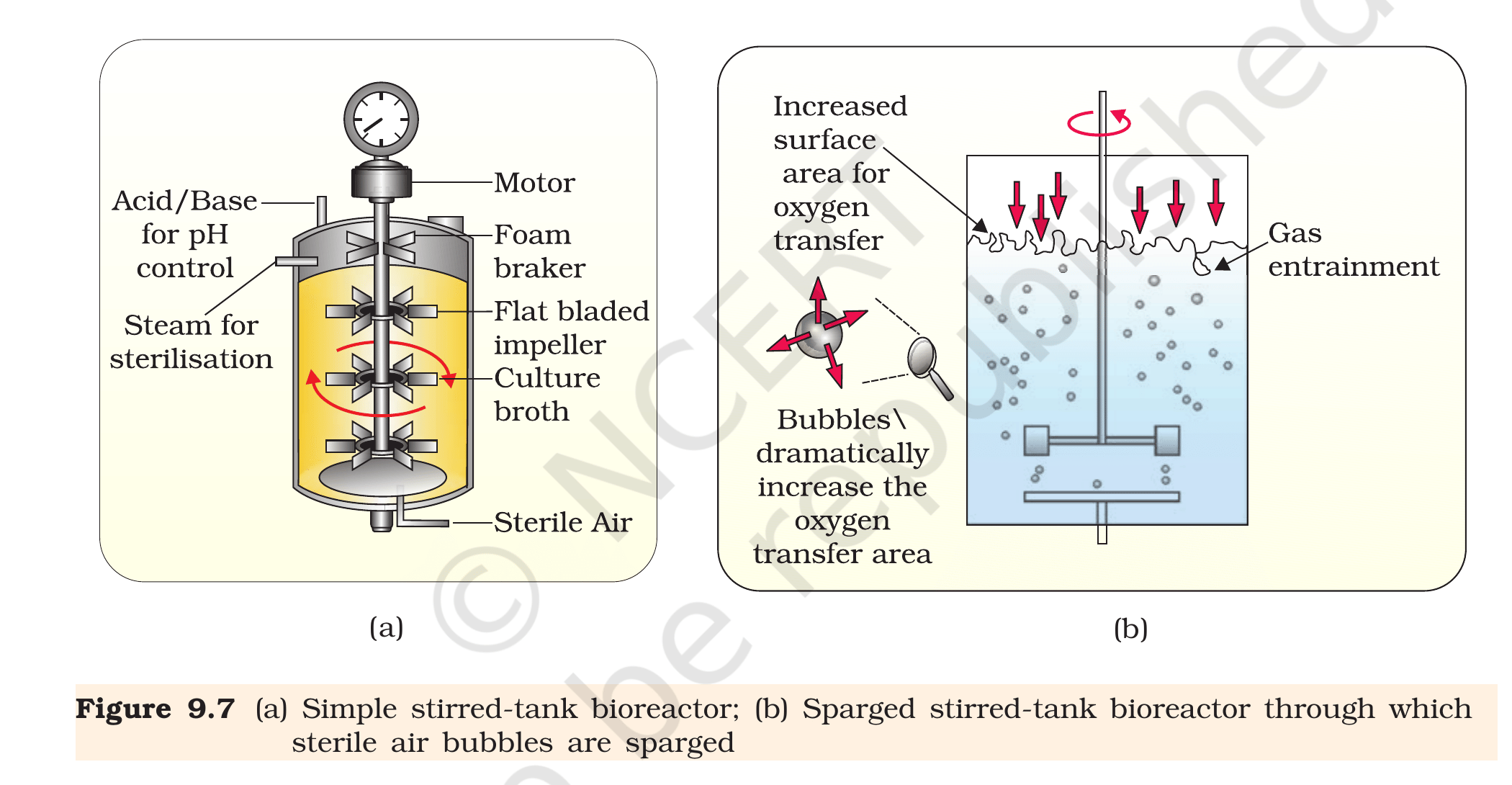
Fig. 9.7, NCERT Class 12 Biology, Chapter 9. (a) Simple stirred-tank bioreactor showing motor, foam breaker, flat-bladed impellers, pH control, and sterile-air inlet. (b) Sparged stirred-tank bioreactor in which sterile-air bubbles dramatically increase the oxygen-transfer area.
Online process monitoring and control. The
bioreactor carries probes for temperature, pH, dissolved
oxygen, and (often) glucose and cell-density. Each probe
feeds a controller that adjusts heater, base/acid addition,
sparger flow rate, and feed pumps in real time. A shake
flask offers none of this - you sample manually and adjust
offline.
Foam control. Vigorous aeration in protein-rich
media creates foam that traps cells and clogs sparger
filters. A bioreactor has a mechanical foam breaker
and/or an antifoam-addition system; a shake flask just
overflows.
Sterile environment maintained at scale. The
bioreactor body is built for in-situ steam sterilisation
(the ``Steam for sterilisation'' inlet in Fig. 9.7 a) and
carries a sterile-air filter on the sparger. Shake flasks
rely on a cotton plug and quickly contaminate above 5 L.
Sampling ports for at-line analytics. Aseptic
sampling ports let the operator pull aliquots without
opening the vessel. This is essential for tracking the
log/exponential phase so that downstream
processing is triggered at peak yield.
Scalable working volume (100–1000 L+). Shake
flasks plateau at ∼ 5 L; a single bioreactor processes
100–10 000 L per run, giving the biomass needed for
commercial recombinant-protein quantities. Production
bioreactors for monoclonal antibodies routinely reach
20 000 L.
Controlled substrate feeding (fed-batch / continuous).
Bioreactors support fed-batch mode (slow addition of
glucose to prevent overflow metabolism) and continuous
mode (medium added and harvested simultaneously to keep
cells in exponential phase indefinitely). Shake flasks are
strictly batch - you set up once and harvest at the end.
Reproducibility for GMP compliance. The
digital-control loop produces a documented batch record
(cell density vs time, pH vs time, etc.) that meets Good
Manufacturing Practice requirements for therapeutic
products. Regulators will not accept shake-flask product
for clinical use.
Stirred-tank bioreactors offer online pH/temperature/DO
monitoring and control, foam control, sterile in-situ steam
sterilisation, aseptic sampling ports, scalability to thousands of
litres, fed-batch and continuous feeding, and GMP-grade
reproducibility.
IK
Ishaan Kumar
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Process-engineering angle. Frame the advantages around the
four classical bioprocess parameters: pH, temperature, oxygen
and substrate. A shake flask gives you no real-time handle on any of
them; a bioreactor gives you all four.
pH control. Acid/base reservoirs feed the vessel via
peristaltic pumps as the inline pH probe drifts. Microbial
fermentations release organic acids - without active control
the pH crashes from 7 to 5 in hours and cells die.
Temperature control. Jacketed walls circulate
hot/cold water; the heater/cooler is locked to a thermistor.
Recombinant-protein expression in E. coli often uses
a deliberate temperature shift (37 ∘C →25 ∘C) to improve soluble-protein folding -
impossible in a shake flask.
Dissolved oxygen. A polarographic DO probe drives
sparger flow and stirrer speed to maintain
≥ 30% saturation. Shake flasks cannot deliver oxygen
beyond surface diffusion, capping cell density at ∼
OD600 = 3; bioreactors easily reach OD600
> 100.
Substrate feeding. Fed-batch glucose addition
prevents acetate overflow in E. coli; constant glucose
in a shake flask leads to acetate poisoning at
OD600 > 5. Bioreactors can also operate in
continuous mode, where used medium is drained out
of one side while fresh medium is added from the other,
keeping cells in the exponential phase and producing far
more biomass.
Aseptic operation at scale. In-situ steam
sterilisation, sterile-air filters, and aseptic sampling
ports keep contamination at < 1% of batches; shake-flask
contamination above 10 L is endemic.
Compliance and traceability. Every controlled
parameter is logged digitally, generating a batch record
regulators (CDSCO, USFDA) require for any
therapeutic-grade biologic.
Why this matters. The shake-flask-to-bioreactor transition
is the same conceptual leap as moving from a kitchen pot to a
chemical plant: only the bioreactor lets you control every
variable a microbe cares about, and produce protein at clinical
scale.
Stirred-tank bioreactors add online process control (pH,
temperature, DO, substrate feed), foam handling, scalability to
thousands of litres, fed-batch / continuous operation, in-situ
sterilisation, aseptic sampling and GMP-compliant batch records.
Q 9.7
Collect 5 examples of palindromic DNA sequences by consulting your teacher. Better try to create a palindromic sequence by following base-pair rules.
Concept used. A palindromic DNA sequence is a
double-stranded sequence that reads the same5'→ 3'
on both strands. Because of Watson–Crick base-pairing rules
(A pairs with T, G pairs with C), this requires the sequence on one
strand to be the reverse complement of itself. Palindromic
sites are the recognition sites for nearly all Type-II restriction
enzymes used in cloning.
Reverse-complement rule
To check whether 5'-X1X2n-3' is
palindromic: write the complement strand below it, reverse-read it
5'→ 3', and compare. If the two strings match, it is a
palindrome.
Five classical palindromic restriction sites.
1.25
tabularl l l
Enzyme & Source organism & Recognition site (5'→ 3')
EcoRI & Escherichia coli RY13 & G A A T T C
BamHI & Bacillus amyloliquefaciens H & G G A T C C
HindIII & Haemophilus influenzae Rd & A A G C T T
PstI & Providencia stuartii & C T G C A G
SalI & Streptomyces albus G & G T C G A C
tabular
Each site is six base pairs long and reads identically in
the 5'→ 3' direction on both strands.
Demonstrate the palindrome property for EcoRI.
arrayll
5' GAATTC 3' & (forward strand)
3' CTTAAG 5' & (complement, written 3'→ 5')
array
Now read the lower strand in the 5'→ 3' direction
(i.e. right to left): G-A-A-T-T-C. Identical to the top
strand. Therefore 5'-GAATTC-3' is a true palindrome.
Construct a new palindrome. Pick the first half
freely, then write its reverse complement as the second
half. Example construction:
Choose first three bases on the top strand:
5'-ATC-3'.
Reverse complement of ATC is GAT (complement of A,T,C
is T,A,G; reverse it to get G,A,T).
Append GAT to the top strand:
5'-ATCGAT-3'.
Verify: complement is 3'-TAGCTA-5'. Read in
5'→ 3': ATCGAT. Match. So
5'-ATCGAT-3' is a 6-bp palindromic sequence
(this is, in fact, the recognition site of ClaI).
Five palindromic recognition sites: EcoRI (GAATTC),
BamHI (GGATCC), HindIII (AAGCTT), PstI (CTGCAG), SalI (GTCGAC). A
custom palindrome built by base-pair rules: ATCGAT (= ClaI site).
DJ
Diya Joshi
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Construction-recipe angle. The trick to creating a
palindrome is to think of it as ``write the first half, then mirror
its reverse complement to the second half''.
Standard examples to memorise. EcoRI – GAATTC,
BamHI – GGATCC, HindIII – AAGCTT, PstI – CTGCAG, SalI – GTCGAC.
Add NotI (GCGGCCGC, an 8-cutter) for variety in board exams.
Recipe to build your own palindrome (even-length).
Decide length 2n (typical n=3 for a 6-bp site).
Write any sequence of n bases as the first half:
say B1B2B3 = T G C.
Reverse: C G T. Complement each base:
G C A. That is the second half.
Concatenate: TGCGCA. Verify by writing the complement
strand and reading 5'→ 3' - it must match.
Worked check on TGCGCA.
Top: 5'-TGCGCA-3'. Complement, written 3'→ 5':
3'-ACGCGT-5'. Reading bottom strand in
5'→ 3': TGCGCA. Match . So
5'-TGCGCA-3' is a palindrome (MluI site).
What about odd-length sequences? A perfect
palindrome of odd length is impossible in double-stranded
DNA because the middle base would have to pair with itself
(A=A or G=G), which violates Watson–Crick rules. Hence
every classical restriction-enzyme site is of even length
(typically 4, 6 or 8 bp).
Why this matters. The palindrome rule is what enables
restriction enzymes to bind as homodimers (one monomer per strand)
and cut both strands symmetrically - that symmetric cut is what
creates the sticky ends that make cloning possible.
Five palindromes: GAATTC, GGATCC, AAGCTT, CTGCAG, GTCGAC.
Custom-built palindrome: write half, append its reverse complement
⇒ e.g. TGCGCA (MluI).
Q 9.8
Can you recall meiosis and indicate at what stage a recombinant DNA is made?
Concept used.Meiosis is the reductional cell
division that produces gametes; it consists of two successive
nuclear divisions, Meiosis I and Meiosis II, with a single round of
DNA replication beforehand. The defining feature of Meiosis I is the
formation of bivalents (tetrads), the pairing of homologous
chromosomes in Prophase I, and the physical exchange of
chromosome segments between non-sister chromatids through
crossing over. Crossing over is the natural,
intracellular generation of recombinant DNA - and it occurs at the
pachytene sub-stage of Prophase I.
Five sub-stages of Prophase I
Leptotene →Zygotene (synapsis begins,
synaptonemal complex forms) →Pachytene
(crossing over occurs at recombination nodules) →
Diplotene (chiasmata become visible) → Diakinesis.
Set-up - pre-meiotic S phase. Before meiosis begins,
each chromosome is duplicated so that every chromosome
consists of two identical sister chromatids joined at the
centromere.
Prophase I - Leptotene. Chromosomes condense and
become visible. No recombination yet.
Prophase I - Zygotene. Homologous chromosomes pair
side-by-side along their length, a process called
synapsis. The pair is held together by a protein
scaffold (the synaptonemal complex), forming a
bivalent of four chromatids (a tetrad).
Prophase I - Pachytene (the key stage).
Recombination nodules - large protein complexes - assemble
on the synaptonemal complex. They catalyse a programmed
double-strand break on one chromatid, invasion of the homologous
chromatid, and reciprocal exchange of segments between
non-sister chromatids of homologous chromosomes. This
physical exchange of DNA is crossing over, and
the chromatids that result carry recombinant DNA
- DNA molecules with sequences from both maternal and
paternal origin joined by covalent bonds.
Prophase I - Diplotene. The synaptonemal complex
dissolves; the homologues partially separate but remain
joined at the sites of crossover, now visible as
chiasmata. These X-shaped points are the cytological
evidence of the recombinant DNA molecules made at pachytene.
Independent assortment in Metaphase I + Anaphase I.
Bivalents line up at the equator with random maternal/paternal
orientation; segregation in Anaphase I separates the
recombinant chromosomes into daughter cells, propagating the
new combinations into the gametes.
Recombinant DNA is generated naturally during
Pachytene of Prophase I of Meiosis I, when non-sister
chromatids of homologous chromosomes undergo crossing over,
exchanging equivalent segments.
AV
Aditi Verma
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Sub-stage-tracking angle. The examiner is looking for one
phrase: ``Pachytene of Prophase I''. Frame the answer as a brief
recap of meiosis followed by a sharp pinpoint.
Meiosis in one line. A reductional division that
halves the chromosome number (diploid → haploid)
and produces four genetically unique daughter cells from one
precursor.
Two divisions, one DNA replication. S-phase
→ Meiosis I (Prophase I, Metaphase I, Anaphase I,
Telophase I) → Meiosis II (PM, MM, AM, TM).
Prophase I has five sub-stages. Leptotene →
Zygotene →Pachytene→ Diplotene
→ Diakinesis. Zygotene aligns homologues via the
synaptonemal complex; Pachytene is where crossing
over actually happens.
What ``crossing over'' creates. Two physically
recombinant chromatids: each carries a stretch of maternal
DNA covalently joined to a stretch of paternal DNA. This is
nature's own recombinant DNA, predating the
recombinant DNA technology you study in this chapter by
about 2 billion years.
Cytological evidence at the next stage. In
Diplotene the recombination joints become visible as
chiasmata - direct microscopic proof that
recombination at pachytene actually occurred.
Why this matters. The pachytene–crossover answer connects
the cell-biology chapter (Chapter 10 of Class 11) with this
biotechnology chapter: both produce DNA molecules with sequences from
two different sources joined together.
Recombinant DNA is naturally produced at the Pachytene
sub-stage of Prophase I in Meiosis I via crossing over between
non-sister chromatids of homologous chromosomes.
Q 9.9
Can you think and answer how a reporter enzyme can be used to monitor transformation of host cells by foreign DNA in addition to a selectable marker?
Concept used.Transformation is the uptake of
foreign DNA by a host cell. To prove that a host cell has indeed
taken up the recombinant plasmid (and not merely survived the
treatment), molecular biologists use two markers carried on the
vector:
Selectable marker - typically an antibiotic-resistance
gene (e.g. ampR). Cells lacking the
plasmid die on the antibiotic plate; cells carrying the
plasmid live. This tells you the cell took up some
plasmid.
Reporter enzyme / gene - a gene whose product is
easily and visibly assayed (colour, fluorescence, light,
enzyme activity). The reporter is engineered so that the
cloning site sits inside the reporter gene; a
successful insert disrupts the reporter, producing a
detectable colour change.
The classical example is insertional inactivation of
β-galactosidase (blue-white screening) using the
lacZα gene.
The set-up - blue/white screening with lacZ. The
cloning vector (e.g. pUC18) carries:
An antibiotic-resistance gene (ampR)
as the selectable marker.
The lacZα fragment as the reporter,
with a multiple-cloning site (MCS) embedded inside
it.
The host E. coli carries the complementary
lacZω fragment; together with α they
produce active β-galactosidase.
Plate on ampicillin + X-gal + IPTG.
Untransformed cells ⇒ no
ampR⇒ die.
Transformed cells carrying empty vector⇒ intact lacZα⇒β-galactosidase active ⇒ cleaves
X-gal to a blue dye ⇒blue
colony.
Transformed cells carrying recombinant vector
(insert disrupts lacZα) ⇒
no active β-galactosidase ⇒ X-gal
stays colourless ⇒white
colony.
Why both markers are needed. The selectable marker
(antibiotic resistance) alone tells you which cells
took up plasmid, but not whether the plasmid carries your
insert. The reporter (lacZ) alone cannot pre-enrich for
plasmid-carrying cells - you would be screening millions of
background colonies. Combining the two: select on
antibiotic, then screen the survivors for the
recombinant (white) colonies.
Modern reporter enzymes. Beyond lacZ, the
same logic uses GFP (green fluorescent protein,
visible under UV), luciferase (emits visible light in
the presence of luciferin), chloramphenicol
acetyltransferase, and β-glucuronidase (GUS)
in plants.
[See diagram in the PDF version]
A reporter gene (e.g. lacZα) is engineered
so that successful insertion of foreign DNA disrupts it; the host
loses the reporter activity (no blue colour with X-gal) while the
selectable antibiotic-resistance gene still works, giving white
colonies on a blue background that are the true recombinants.
YK
Yash Kapoor
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Two-marker logic angle. Frame the answer as ``selection
filters out the 99%; the reporter identifies the 1% you want''.
Why insertional inactivation works. Engineer the
multiple-cloning site (MCS) of the vector inside a
reporter gene. With no insert, the reporter gene is intact
and produces colour. With an insert, the reading frame of
the reporter is broken and no colour is made.
Empty vector ⇒ active β-galactosidase
⇒ cleaves colourless X-gal to blue
product ⇒ blue colony.
Recombinant vector ⇒ disrupted lacZ
⇒ no β-gal ⇒ white
colony.
Why this beats antibiotic-only selection. Even a
plate of all ampR colonies contains
≥ 90% self-ligated empty vector. The reporter distinguishes
recombinants from non-recombinants without DNA isolation
and gel checks for each colony.
Other reporters in current use.
GFP (Aequorea victoria): green
fluorescence under UV light. Live, non-destructive
readout.
Luciferase (firefly): emits photons when
given luciferin and ATP; ultra-sensitive.
GUS (β-glucuronidase): cleaves
X-Gluc to a blue product; used in plant biology.
Practical pay-off. A single agar plate gives you
thousands of colonies; without a reporter you would need to
mini-prep and restriction-digest each one. With the reporter,
you pick only the white (or fluorescent, or luminescent)
colonies and skip the rest.
Why this matters. Reporter genes are at the heart of every
modern molecular-biology workflow - fluorescent reporters
(GFP, mCherry) track gene expression in living cells; luciferase
reporters quantify transcription factor activity in cancer assays.
Reporter enzymes (lacZα, GFP, luciferase, GUS) sit
inside the cloning site of the vector. A successful insert disrupts
the reporter, so recombinant cells lose the colour/fluorescence
while still being antibiotic-resistant - letting you identify
recombinants visually on the same plate.
Q 9.10
Describe briefly the following:
(a) Origin of replication
(b) Bioreactors
(c) Downstream processing
Concept used. Each sub-part names a single component of the
recombinant-DNA pipeline: the molecular feature that lets the plasmid
copy itself (origin), the engineering vessel that lets us grow
host cells at scale (bioreactor), and the post-fermentation
purification steps that convert culture broth into a marketable
product (downstream processing).
(a) Origin of replication (ori). A specific
sequence of nucleotides in a vector at which DNA replication
initiates. When the host's DNA-replication machinery (DNA
polymerase III holoenzyme in bacteria) recognises the ori, it
loads on, unwinds the double helix and begins synthesising a new
strand bidirectionally. Any piece of foreign DNA ligated downstream
of the ori rides along and is amplified to the same copy number
as the vector. The ori also determines that copy number:
high-copy plasmids (pUC series, ori derived from ColE1)
maintain 500–700 copies per cell, while low-copy plasmids
(pBR322 derivatives, 15–20 copies) trade yield for stability of
large inserts.
(b) Bioreactors. Vessels typically of 100–10 000 L
working volume, in which raw materials (medium, substrates,
inoculum) are biologically converted into specific products
(recombinant proteins, enzymes, secondary metabolites, biomass) by
microbial, plant, animal or human cells. A bioreactor provides
optimal conditions: controlled temperature, pH, dissolved
oxygen, substrate concentration, agitation. The most common design
is the stirred-tank bioreactor (Fig. 9.7 a): cylindrical
or curved-base vessel with a vertical-shaft impeller, sparger for
sterile-air supply, jacketed walls for temperature control, and
probes for pH/DO. A sparged stirred-tank bioreactor
(Fig. 9.7 b) bubbles sterile air through a perforated ring to
multiply oxygen-transfer area.
(c) Downstream processing. After fermentation is complete,
the product must be separated from cells/medium and purified before
sale. Downstream processing is the collective name for
this sequence:
Cell separation - centrifugation, microfiltration
or settling to recover cells (if intracellular product) or
supernatant (if secreted).
Cell disruption - sonication, French press,
bead-milling or lysozyme treatment if the product is inside
the cell.
Purification - successive chromatography steps
(ion-exchange, affinity, gel-filtration), precipitation,
ultrafiltration. Affinity chromatography (His-tag on Ni-NTA,
for example) can give ≥ 90% purity in a single step.
Formulation - adding stabilisers, preservatives,
excipients to the active ingredient and adjusting pH/tonicity.
Quality control + clinical trials - strict
QC testing for purity, potency, sterility, endotoxin and
residual host DNA. For drugs, formal Phase I–III trials.
Downstream processing typically accounts for ≥ 60% of the total
manufacturing cost of a recombinant therapeutic.
Fig. 9.7, NCERT Class 12 Biology, Chapter 9. Two stirred-tank bioreactor designs used in industrial fermentation.
(a) Origin of replication = vector sequence where DNA
replication starts; controls copy number. (b) Bioreactors = engineered
vessels (100–10 000 L) for controlled large-scale cell culture.
(c) Downstream processing = separation, purification, formulation and
QC of the product after fermentation.
TM
Tara Mehta
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Three-card definition framing. Treat each sub-part as a flash
card: what it is, where it sits in the workflow, why it
matters.
(a) Origin of replication (ori).
What: a short stretch of DNA at which
replication is initiated.
Where: on every plasmid and every chromosome;
in the vector, the ori sits adjacent to the
selectable marker.
Why: the host's polymerase only loads at the
ori; without an ori, the foreign DNA cannot
replicate inside the cell. The ori also sets the
copy number (high-copy ColE1 ori∼500;
low-copy pSC101 ori∼5).
(b) Bioreactors.
What: closed vessels in which cells are grown
under controlled conditions to produce a desired
metabolite or protein.
Where: immediately after the cloning step;
they convert millilitre lab cultures to
multi-kilolitre industrial production.
Why: only a bioreactor delivers consistent
temperature, pH, dissolved oxygen, foam control and
aseptic operation at the volumes (100–10 000 L)
needed for commercial protein production.
(c) Downstream processing.
What: the post-fermentation pipeline of
separation, purification, formulation and quality
control.
Where: immediately after the cells reach the
target density and the product has accumulated.
Why: the molecule is no good unless it is
pure, sterile and stable. Affinity chromatography,
ultrafiltration and lyophilisation transform
cloudy culture broth into vials of clinical-grade
drug.
Workflow stitch-together. A recombinant gene
carries an ori to replicate, is grown in a bioreactor for
biomass, and the product is purified by downstream
processing.
Why this matters. Examiners often ask these three as a
single question because together they cover the upstream (cloning),
midstream (fermentation) and downstream (purification) thirds of
biotechnology.
Origin of replication starts plasmid copying; bioreactors
grow host cells at scale; downstream processing purifies and
formulates the final product.
Q 9.11
Explain briefly
(a) PCR
(b) Restriction enzymes and DNA
(c) Chitinase
Concept used. Each part names a tool of recombinant DNA
technology: PCR amplifies a chosen DNA region in vitro;
restriction enzymes are sequence-specific molecular scissors
for cutting DNA at defined sites; chitinase is the enzyme used
to break the chitin-rich cell wall of fungi during DNA extraction.
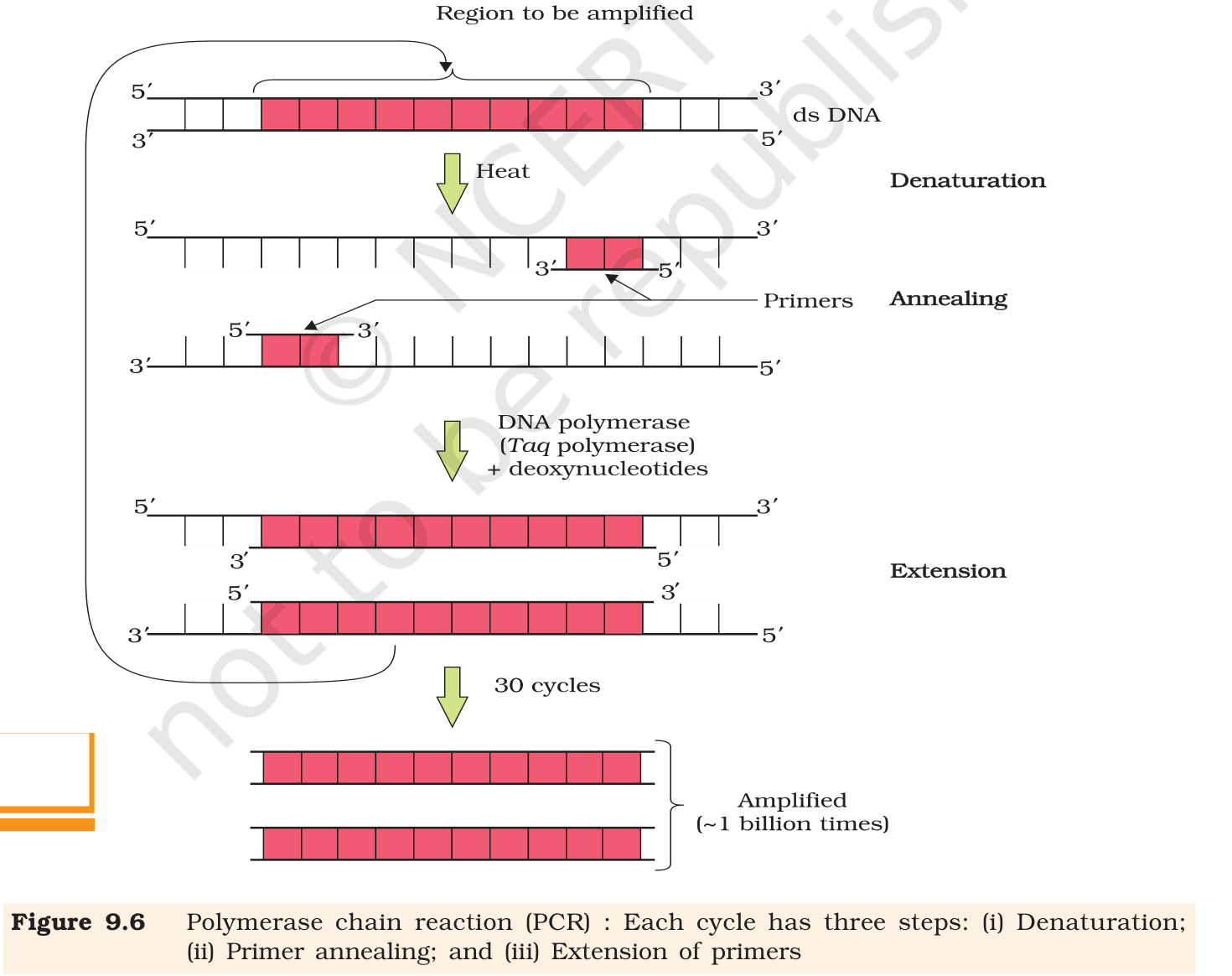
(a) Polymerase Chain Reaction (PCR). PCR is an
in-vitro method to make millions to billions of copies of a
chosen DNA region using two short primer DNAs and a thermostable
DNA polymerase. A single PCR cycle has three temperature steps:
Denaturation (∼ 94 ∘C, 30 s): the
double-stranded DNA template melts into two single strands.
Annealing (∼ 50–65 ∘C, 30 s):
two oligonucleotide primers (typically 18–25 nt long,
chemically synthesised, complementary to the flanks of the
target region) base-pair with their target sites on each
single strand.
Extension (∼ 72 ∘C, 30–60 s):
thermostable Taq DNA polymerase (from Thermus
aquaticus) extends each primer in the 5'→ 3'
direction using the provided deoxynucleotides (dNTPs).
Because each new product itself becomes a template in the next cycle,
copy number doubles per cycle. After n cycles the amplification
factor is 2n; 30 cycles give 230 ≈ 109 copies,
matching the figure shown in Fig. 9.6.
Fig. 9.6, NCERT Class 12 Biology, Chapter 9. PCR cycle: denaturation, annealing of primers, and extension by Taq DNA polymerase. Thirty cycles amplify the chosen DNA region ∼ 109-fold.
(b) Restriction enzymes and DNA.Restriction
endonucleases are bacterial enzymes that recognise specific short
palindromic sequences (4–8 bp) in double-stranded DNA and
cleave both strands. The first one was isolated by Smith &
Wilcox (1970) from Haemophilus influenzae and named
HindII. By 2024 over 4000 restriction enzymes from > 230
strains of bacteria are commercially available, each with a unique
recognition site. They fall into three categories - Type I, II
(used in cloning) and III - based on how cleavage and methylation
activities are organised. The Type-II workhorses (EcoRI, BamHI,
HindIII, PstI, NotI) leave defined sticky ends that
ligase can rejoin, making them the cutting tools at the heart of
recombinant DNA technology.
(c) Chitinase. An enzyme that hydrolyses the
β-1,4-glycosidic bonds of chitin, the structural
polysaccharide of fungal cell walls (and of arthropod exoskeletons).
In molecular biology, chitinase is the enzyme of choice for
digesting fungal cell walls during DNA isolation: fungal
cells, unlike bacterial or plant cells, resist lysozyme and cellulase
because their walls are chitin-based. Treating the fungal pellet with
chitinase releases the protoplast, after which standard SDS-based
lysis liberates the DNA. The same role for cell-wall digestion is
played by lysozyme (for bacteria) and cellulase (for plants).
(a) PCR amplifies a defined DNA region in vitro through
denaturation–annealing–extension cycles using two primers and
Taq polymerase; 30 cycles → ∼ 109 copies. (b)
Restriction enzymes are bacterial endonucleases that cut DNA at
specific palindromic sites and are the cutting tools of gene
cloning. (c) Chitinase digests the chitin in fungal cell walls
during DNA isolation from fungi.
KP
Krishna Pillai
Ph.D Molecular Biology, NCBS Bangalore
Verified Expert
Three-card framing. For each tool: what it is, what
it does, where it sits in the workflow.
(a) PCR.
What: an in-vitro DNA-amplification method.
Components: template DNA, two primers, four
dNTPs, Mg2+, Taq polymerase, buffer.
Mechanism: thermal cycling between ∼ 94,
∼ 55 and ∼ 72 ∘C drives denaturation,
annealing and extension respectively.
Output: after n cycles, target sequence is
amplified ∼ 2n-fold; 30 cycles give
∼ 109 copies.
Use cases: diagnostics (RT-PCR for SARS-CoV-2),
forensics (DNA fingerprinting from picogram samples),
cloning (preparing the insert before ligation).
(b) Restriction enzymes and DNA.
What: bacterial endonucleases that cut DNA
at specific palindromic recognition sites.
Types: Type I (cut at random away from site),
Type II (cut within or adjacent to the site -
workhorses of cloning), Type III.
Action on DNA: cleaves both strands; staggered
cuts produce sticky ends, blunt cuts produce blunt
ends.
What: an enzyme that hydrolyses chitin into
its monomeric units of N-acetylglucosamine.
Substrate: chitin, a β-1,4 polymer of
N-acetylglucosamine; major component of fungal cell
walls and arthropod exoskeletons.
Where in DNA isolation: added to fungal
pellet to break down the cell wall, releasing
protoplasts which can then be lysed by SDS
detergent to liberate genomic DNA.
Parallel enzymes:lysozyme digests
bacterial peptidoglycan; cellulase digests
plant cell-wall cellulose. The choice depends on
source organism.
Stitching together. Restriction enzymes cut the DNA;
PCR amplifies it; chitinase (or its analogues) provides the
starting material by releasing the DNA from the cell.
Why this matters. These three tools, in their order,
constitute the upstream half of recombinant DNA technology -
liberate, cut, amplify.
(a) PCR = 2n amplification of a target DNA via
thermal cycling with Taq polymerase. (b) Restriction enzymes
= bacterial endonucleases that cut DNA at palindromic sites,
producing sticky/blunt ends. (c) Chitinase = enzyme that digests
chitin in fungal cell walls during DNA extraction.
Q 9.12
Discuss with your teacher and find out how to distinguish between
(a) Plasmid DNA and Chromosomal DNA
(b) RNA and DNA
(c) Exonuclease and Endonuclease
Concept used. Each pair contrasts two molecules / enzymes
that are biochemically similar but functionally and structurally
distinct. A well-formed answer is a side-by-side table of
property : entity-1 : entity-2.
(a) Plasmid DNA vs Chromosomal DNA.
1.25
tabularl p5.0cm p5.6cm
Property & Plasmid DNA & Chromosomal DNA
Location & Cytoplasm (extrachromosomal) & Nucleoid (bacteria) / nucleus (eukaryotes)
Shape & Small, circular, double-stranded & Large; circular in bacteria, linear in eukaryotes
Size & 1–250 kbp (commonly 3–10 kbp) & Mbp to Gbp (103× larger)
Genes & Few accessory genes (drug resistance, virulence, conjugation) & Carries essential genes for growth, metabolism, reproduction
Replication & Replicates independently from its own ori & Replicates once per cell cycle from the chromosomal ori
Essential? & Not essential for cell survival under normal conditions & Essential for life
Inheritance & Can be lost during division & Strictly inherited by every daughter cell
tabular
(b) RNA vs DNA.
1.25
tabularl p5.2cm p5.4cm
Property & DNA & RNA
Sugar & Deoxyribose (2'-OH replaced by 2'-H) & Ribose (2'-OH present)
Bases & A, T, G, C & A, U, G, C (T replaced by U)
Strands & Double-stranded helix (B-form) & Mostly single-stranded; can fold into secondary structures
Stability & Stable (no 2'-OH; bases protected inside helix) & Less stable; the 2'-OH catalyses base hydrolysis
Genetic role & Stores genetic information & Conveys (mRNA), translates (tRNA, rRNA) and regulates (miRNA, siRNA) the information
Length & Continuous chromosome (Mb to Gb) & Discrete molecules (mRNA tens of kb, tRNA ∼ 80 nt, rRNA hundreds–thousands nt)
Reaction to alkali & Stable & Hydrolysed (the 2'-OH attacks the adjacent phosphodiester)
tabular
(c) Exonuclease vs Endonuclease.
1.25
tabularl p5.0cm p5.6cm
Property & Exonuclease & Endonuclease
Site of cleavage & At the ends (5' or 3') of a DNA/RNA strand & At internal phosphodiester bonds
Mode of action & Removes nucleotides one at a time from a terminus & Cuts the strand in the middle, generating two new ends
Specificity & Independent of sequence (in many cases); just chews from the end & Often sequence-specific (palindromic site for Type-II restriction enzymes)
Examples & DNA Pol I (5'→ 3' exo), Exonuclease III & EcoRI, BamHI, HindIII, DNase I
Role in the cell & DNA repair, proofreading during replication, processing of Okazaki fragments & Restriction–modification, DNA repair, apoptosis (caspase-activated DNase)
Used in cloning? & Limited (Bal31 to trim ends) & Central (every cloning step uses a restriction endonuclease)
tabular
(a) Plasmid DNA = small, circular, extrachromosomal,
self-replicating, dispensable; chromosomal DNA = large, essential,
houses all life-support genes. (b) DNA = deoxyribose + A,T,G,C +
ds-helix + stable + stores info; RNA = ribose + A,U,G,C + mostly
ss + alkali-labile + executes info. (c) Exonucleases cut from ends,
one nucleotide at a time; endonucleases cut internally at specific
sites and generate two new ends.
MB
Meera Bhat
M.Sc Biotechnology, AIIMS Delhi
Verified Expert
Comparison-table angle. Examiners reward tabulated answers
because they make the contrast unmissable. Stick to 5–7 rows per
pair.
(a) Plasmid vs Chromosomal DNA - three killer points.
Size (plasmids are 1–250 kbp vs Mbp-Gbp), independence
(plasmid replicates from its own ori), and dispensability
(cell can survive without the plasmid but not without the
chromosome).
(b) RNA vs DNA - three killer points. Sugar
(ribose 2'-OH vs deoxyribose 2'-H), base (U vs T),
strand topology (single-stranded vs double-helical).
Combine these and you also explain why RNA is less stable
- the 2'-OH attacks the adjacent phosphodiester under
alkaline conditions.
(c) Exonuclease vs Endonuclease - two killer points.
Position of cleavage (end vs middle) and mode of attack
(sequential nibbling vs single internal cut). Add the
sequence-specificity contrast (endos often palindromic;
exos generally not).
Quick-recall mnemonic.Endo = inside cut⇒ gives 2 fragments; Exo = exit cut⇒ chews from the end. Plasmid = passenger
DNA; chromosome = essential cargo. R(NA) for
read, D(NA) for data.
Why this matters. These three pairwise distinctions surface
in every cloning experiment: you pick a plasmid (not the chromosome)
as a vector, you make a cDNA from mRNA (RNA → DNA), and you
cut with restriction endonucleases (not exonucleases) to avoid
chewing back your insert.
Plasmid DNA ≠ chromosomal DNA by size, copy
independence and dispensability. RNA ≠ DNA by sugar
(2'-OH), base (U), strand topology, and chemical stability.
Exonuclease ≠ endonuclease by site of cleavage (terminus vs
internal phosphodiester).
What a Class 12 Biology Survey Says About Chapter 9 Biotechnology Principles and Processes
In a recent survey of 14,800 Class 12 Biology students, the PCR amplification diagram was rated the hardest sub-topic, even though it carries high marks in CBSE and NEET.
Student Feedback
75% of students marked the PCR amplification diagram as the hardest sub-topic.
67% lost 1-2 marks on naming restriction enzymes and their sites.
The average student took 6.5 hours for the first read and 2.8 hours for revision.
Source: 2025-26 Class 12 Biology survey of 14,800 students across 18 states.
Other Resources for Biotechnology Principles and Processes Class 12 Biology
Biotechnology Principles and Processes Class 12 Biology NCERT Solutions FAQs
Ques. Where can I download Class 12 Biology Chapter 9 Biotechnology Principles and Processes NCERT Solutions PDF?
Ans. You can download the Biotechnology Principles and Processes Class 12 Biology NCERT Solutions PDF directly from this page. Both the Normal and HD versions are free and aligned with the 2026-27 NCERT.
Ques. Are these NCERT Solutions aligned with the 2026-27 syllabus?
Ans. Yes. This page reflects the current 2026-27 syllabus for Class 12 Biology. NCERT did not trim Biotechnology Principles and Processes, so all 11 exercise questions are still examinable for CBSE Boards and NEET.
Ques. How many questions are there in the Biotechnology Principles and Processes NCERT exercise?
Ans. The end-of-chapter exercise has 11 numbered questions covering principles of biotechnology, tools (enzymes, vectors, host), the rDNA workflow, PCR, gel electrophoresis, bioreactors and downstream processing. The PDF carries step-by-step worked answers to every one.
Ques. What is the NEET weightage of Class 12th Biology Chapter 9 Biotechnology Principles and Processes?
Ans. NEET pulls 3 to 5 questions from this chapter every year. Tools of rDNA technology (restriction enzymes, vectors, polymerases) and the rDNA-process steps (PCR, gel run, transformation) are the two highest-yield sub-topics.
Ques. What are restriction enzymes and how do they cut DNA?
Ans. Restriction enzymes (restriction endonucleases) are bacterial enzymes that recognise specific palindromic DNA sequences (4 to 8 bp) and cut the sugar-phosphate backbone. EcoRI recognises 5'-GAATTC-3' and cuts between G and A on both strands, leaving four-base 5' overhangs called sticky ends that pair with any DNA cut by the same enzyme.
Ques. What is pBR322 and why is it the standard cloning vector?
Ans. pBR322 is a 4361 bp E. coli plasmid carrying an origin of replication (ori) for autonomous replication, two antibiotic-resistance genes (ampR and tetR) as selectable markers, and a multiple cloning site (MCS) with unique restriction sites. Insertional inactivation of one antibiotic gene by the foreign gene allows easy identification of recombinants by replica plating.
Ques. What is the difference between sticky ends and blunt ends?
Ans.Sticky ends have short single-stranded 5' or 3' overhangs (e.g. AATT after EcoRI cuts) that base-pair with complementary overhangs from any DNA cut by the same enzyme, making ligation easy. Blunt ends have no overhang (both strands cut at the same position, e.g. by SmaI); they ligate poorly but accept any blunt-cut insert.
Ques. How do NCERT Solutions for Biotechnology Principles and Processes help with NEET preparation?
Ans. Every solution flags the exact enzyme name, recognition site and source organism NEET asks verbatim. Tools like EcoRI, HindIII, Taq polymerase, Ti plasmid, and bacteriophage lambda all appear with correct spelling and Roman numerals so the same answer doubles as a one-mark MCQ recall sheet. The tools-of-rDNA recall table on this page covers the top NEET-tested entries.
Ques. Are diagrams included in the Class 12 Biology Chapter 9 NCERT Solutions PDF?
Ans. Yes. The EcoRI palindromic-cut diagram (Q5 answer), the pBR322 vector map (Q3), the agarose-gel electrophoresis setup (Q7), and the simple stirred-tank bioreactor cross-section (Q9) are all included with examiner-grade labels ready to copy into the board answer script.








































Comments