Chemistry Content Strategist | JEE Mentor, 16 Years | Updated on - May 25, 2026
Amines questions appear in every JEE Main shift, with at least one item per paper on basicity ordering, Hoffmann bromamide or the Hinsberg test, and NEET pulls 2 to 3 questions a year on the same topics. Class 12 Chemistry Chapter 9 Amines is therefore a settled scoring chapter, and the 2026-27 NCERT keeps all 25 representative Exemplar items intact. This page hosts the worked-out PDF and the latest pattern.
CBSE Weightage: 4 to 6 marks (typically a 2-mark VSA on Hinsberg or carbylamine test, a 3-mark SA on aniline preparation or basicity ordering, and a 5-mark LA on identifying A/B/C in a multi-step sequence in alternate years)
JEE Main Weightage: 2 to 3% (about 1 to 2 questions per shift on aqueous vs gas-phase basicity, Sandmeyer products, Gabriel synthesis, and the diazonium coupling reactions)
NEET Weightage: 2 to 3 questions per year, leaning on amine classification, Hoffmann bromamide carbon count, and the aniline vs alkylamine basicity contrast
These Exemplar Solutions are curated by Collegedunia subject experts, mapped to the 2026-27 NCERT, and benchmarked against the last five years of CBSE Board, JEE Main and NEET papers.
Every item in the PDF is solved twice: a Solution that lays out the working, then an Expert's Solution that names the controlling concept, so you collect both the answer and the reasoning shortcut on the same page.
Exemplar-Specific Common Mistakes in Amines That Cost JEE and Board Marks
The Amines Exemplar is famous for trap MCQs that look like basicity-ordering questions but test something else (solvation, steric crowding, or aryl-amine resonance). The list below catalogues the five mistakes Collegedunia faculty see most often, with the impact on score.
Confusing aqueous and gas-phase basicity. In water, two methyls (dimethylamine) win because solvation still works; in the gas phase, three alkyls beat two. Mixing the two orders costs an automatic 1-mark MCQ in JEE every other shift.
Calling tert-butylamine a tertiary amine. "Tert" describes the carbon, not the nitrogen. Count C–N bonds, not branching on the alkyl group.
Forgetting that Hoffmann bromamide drops one carbon. An amide with n carbons gives an amine with n-1. Skipping the carbon count flips your product in a 5-mark LA.
Picking LiAlH4 for aryl-nitro reduction. LiAlH4 stops at the azo or hydrazo stage with Ar-NO2. Use Sn/HCl, Fe/HCl, or H2/Pt for a clean Ar-NH2. This single misread loses a 2-mark MCQ in NEET almost every year.
Using primary amines in Gabriel synthesis for aryl halides. Gabriel synthesis fails for Ar-X because SN2 does not work on aryl carbons. Aniline cannot be made this way.
Watch Out: When the Exemplar question says "in aqueous medium", the order is 2° > 1° ≈ 3° > NH3. When it says "gas phase" or "in vacuum", the order flips to 3° > 2° > 1° > NH3. Reading the medium is half the question.
How Collegedunia's Amines Exemplar Solutions Help You Score Higher
Amines is the chapter where the wrong concept costs you the whole question. Our Exemplar PDF pairs the worked answer with a named-concept tag so the reasoning stays portable across JEE, NEET and the Board paper.
Every Question Type Solved End-to-End: MCQ-I, MCQ-II, SA, Matching and Assertion-Reason / LA, each with the Solution plus the Expert's Solution.
Concept Stack Named: +I and -I effects, lone-pair resonance into the aryl ring, solvation of ammonium ions, carbon-count rule for Hoffmann, and the Hinsberg solubility test.
JEE and NEET Bridge: Items on basicity ordering, Sandmeyer products, and Gabriel synthesis are tagged with the year they reappeared in a shift paper.
2026-27 Aligned: The new edition keeps Chapter 9 and every Exemplar item; nothing was dropped from this chapter in rationalisation.
Amines Top Time-Per-Question Budget for the Exemplar Set
The 25 Exemplar items split unevenly across types. The budget below comes from Collegedunia's mock-paper sittings and lets you decide between a single-evening attempt and a three-pass plan.
Question Type
Avg Time per Item
Total for Chapter
What Eats the Time
MCQ-I (single-correct)
60 to 90 seconds
~10 min
Confirming the aqueous vs gas-phase order and ruling out trap distractors
MCQ-II (multiple-correct)
2 to 3 minutes
~6 min
Checking every option against the +I, -I, and solvation rules
SA (Short Answer, 2-3 marks)
4 to 6 minutes
~30 min
Drawing the product structure and naming the reagent
Matching / Assertion-Reason
3 to 4 minutes
~12 min
Mapping reagent to product or testing whether the Reason explains the Assertion
LA (Long Answer, 5 marks)
8 to 10 minutes
~25 min
Multi-step A/B/C identification with stereo and regio control
A first-pass solo attempt of the chapter takes about 80 minutes; a second pass with the Expert's Solution next to you takes another 40. That is roughly the budget Collegedunia recommends a week before the Board paper.
Amines Class 12th: Sample MCQ-II Solved with Multiple-Correct Walk-Through
MCQ-II is the type that bleeds marks because students stop after finding one correct option. Below is a fully worked Exemplar-style MCQ-II from the Amines bank that shows the elimination logic in full.
Question. Which of the following statements about aniline (C6H5NH2) are correct?
(i) Aniline is a weaker base than methylamine in aqueous medium.
(ii) The lone pair on the nitrogen of aniline is fully localised on N.
(iii) Aniline gives a positive carbylamine test on heating with CHCl3 and alcoholic KOH.
(iv) Aniline does not undergo Friedel-Crafts acylation directly because the Lewis acid AlCl3 binds the lone pair on N.
Correct options: (i), (iii) and (iv).
Why (i) is correct. In aniline the N lone pair delocalises into the benzene ring, lowering its availability for protonation. Methylamine's lone pair stays on N and is boosted by the +I methyl, so methylamine wins in water.
Why (ii) is wrong. Resonance structures (C6H5-NH2 ↔ ortho/para C-NH2+) explicitly show partial double-bond character to N; the lone pair is delocalised, not localised.
Why (iii) is correct. Aniline is a primary amine; the carbylamine reaction is a diagnostic test for any 1° amine, aliphatic or aromatic.
Why (iv) is correct. The N lone pair on aniline binds AlCl3 to form a complex, deactivating the ring; that is why we protect with acetylation (acetanilide) before Friedel-Crafts.
Remember: In an MCQ-II, marking one correct option and stopping costs you the full mark. The Exemplar penalises partial selection differently from CBSE; read the marking rubric printed on the cover page before you attempt.
Amines Exemplar Sample Assertion-Reason Solved with Full Logic
Assertion-Reason items reward students who can keep two truth values separate. Below is one Amines A-R item solved end-to-end, the way the Exemplar marking scheme expects.
Assertion (A): Aniline cannot be prepared by the Gabriel phthalimide synthesis.
Reason (R): The Gabriel synthesis uses an SN2 reaction of phthalimide potassium salt with an alkyl halide, and aryl halides do not undergo SN2.
Options: (a) Both A and R are true; R is the correct explanation of A. (b) Both A and R are true; R is not the correct explanation of A. (c) A is true, R is false. (d) A is false, R is true.
Correct option: (a).
Verifying A. Aniline is C6H5NH2; the C–N bond would have to form on an aryl carbon. Standard Gabriel synthesis cannot make this bond, so A is true.
Verifying R. Phthalimide potassium attacks the alkyl halide at the saturated carbon via backside SN2. Aryl halides have sp2 carbons and the C–X bond is partial-double-bond by resonance; SN2 does not occur. R is also true.
Linking A and R. The failure of Gabriel synthesis for aniline is exactly because aryl halides resist SN2, which is what R states. So R explains A; option (a).
The 25 representative items in the Collegedunia PDF span all five Exemplar buckets. The table shows the split and the marks each type is worth in a typical Board sitting.
Question Type
Item Range
Count
Typical Marks (Board)
MCQ-I (single-correct)
Q 9.1 to 9.10
10
1 mark each
MCQ-II (multiple-correct)
Q 9.11 to 9.13
3
1 to 2 marks each
Short Answer (SA)
Q 9.14 to 9.19
6
2 to 3 marks each
Matching
Q 9.20 to 9.21
2
3 to 4 marks each
Assertion-Reason / LA
Q 9.22 to 9.25
4
1 to 5 marks each
SA items are the highest-yield: six questions, all worked in the PDF with named concepts, covering preparation routes, basicity, and the named tests.
Diazonium Chemistry Exemplar Drill: Sandmeyer, Gattermann, Schiemann and Azo Coupling
Roughly 30 per cent of the Amines Exemplar items pivot on a diazonium reaction. The five-row matrix below collects the conversions you must recognise on sight; the Exemplar trap is usually a swapped catalyst or a missed temperature.
Conversion
Reagent / Conditions
Product
Exemplar Trap
Diazotisation
NaNO2 + HCl, 273-278 K
Ar-N2+Cl-
Above 278 K the salt hydrolyses to phenol; writing "room temperature" loses the question
Sandmeyer (ArCl, ArBr, ArCN)
CuCl/HCl, CuBr/HBr, CuCN/KCN
Aryl halide / aryl nitrile
Cu(I) salt - swap to Cu powder = Gattermann (lower yield)
Gattermann (ArCl, ArBr)
Cu powder + HCl or HBr
Aryl halide
Distractor option will list "CuCl" - that is Sandmeyer
The Exemplar pattern is consistent: list four reagent-product pairs and ask "which set is correct". Lock the catalyst-temperature-medium triplet for every diazonium reaction, then the matching becomes mechanical.
Amines Exemplar Drill on Aniline Reactions and EAS Selectivity
Aniline EAS is the second-densest sub-topic in the Exemplar bank. The Exemplar tests whether you can predict the regiochemistry (ortho, para, meta) under each condition.
Aqueous Br2 on aniline gives 2,4,6-tribromoaniline as a white precipitate; the -NH2 group is so strongly activating that mono-substitution is not possible. Selecting "p-bromoaniline" as the direct product is a 1-mark trap.
Controlled p-bromoaniline demands the protect-brominate-deprotect route: acetylate to acetanilide, brominate in CH3COOH, hydrolyse. The acetyl group reduces ring activation enough to give clean para-substitution.
Nitration of aniline with conc. HNO3/H2SO4 gives roughly 47% m-nitroaniline because the strong acid protonates -NH2 to -NH3+, the anilinium ion, which is a meta-director. The Exemplar distractor lists "p-nitroaniline" as the major product.
Sulphonation at 453-473 K gives p-sulphanilic acid as a zwitterion. This is the cleanest direct EAS on aniline without prior protection.
Friedel-Crafts alkylation/acylation fails entirely because AlCl3 binds the N lone pair and deactivates the ring; the Exemplar fix is acetylation first.
Remember: The anilinium ion (Ar-NH3+) is a meta-director; neutral aniline (Ar-NH2) is an ortho-para director. The reaction medium controls which one is present, which is exactly what the Exemplar tests in its EAS items.
Amines Exemplar Step-Up from the NCERT Textbook
The NCERT textbook gives the rules; the Exemplar twists them. The table contrasts a textbook setup with the matching Exemplar setup so you can see exactly what the difficulty step adds.
Concept
NCERT Textbook
Exemplar Twist
Basicity ordering
State the order in water
Compare aqueous and gas-phase orders for the same set, asking which option holds in both
Gabriel synthesis
Show synthesis of methylamine
Ask why the same route fails for aniline; require an SN2 / sp2-carbon argument
Hoffmann bromamide
Convert R-CONH2 to R-NH2
Provide a 5-carbon amide and ask for the 4-carbon amine; track the carbon count
Aniline reactions
Mark aniline as ortho/para directing
Ask why direct Friedel-Crafts fails (AlCl3 binds N) and why acetylation is needed
Sandmeyer reaction
List Ar-N2+ reagents
Ask which Cu(I) salt gives Ar-Cl vs Ar-Br vs Ar-CN and predict by-products
The pattern is consistent: NCERT trains the rule, the Exemplar tests whether you can spot the exception. Reading both books in parallel covers both sides.
Best Way to Use the Amines Exemplar for JEE and NEET Prep
Amines yields one JEE Main question every shift and 2 to 3 NEET questions a year. Collegedunia's recommended attempt sequence below is built around that volume.
First Pass (Day 1): Read NCERT Chapter 9 once, write down the four named reactions (Gabriel, Hoffmann, Sandmeyer, Hinsberg). Skip the Exemplar for now.
Second Pass (Day 2): Attempt the 10 MCQ-I and 3 MCQ-II from the Exemplar without looking at the solution. Mark wrong ones for re-attempt.
Third Pass (Day 3): Work the 6 SA and 4 A-R / LA items with the Solution open. Tag each one with its named concept (basicity, carbon count, SN2 failure, etc.).
Fourth Pass (Day 4): Re-attempt only the items you got wrong. Read the Expert's Solution before checking your answer this time.
Revision Card (1 day before exam): Skim only the basicity orders, the Hoffmann carbon-count rule, and the Hinsberg solubility table.
All NCERT Exemplar Questions for Amines with Step-by-Step Solutions
Every question of the NCERT Exemplar set for Class 12 Chemistry Chapter 9 Amines is listed below with its full Solution and Expert Solution hidden inside collapsible tabs. Click Check Solution to reveal the step-by-step working; click Expert Solution for the expanded explanation.
I. Multiple Choice Questions (Type-I)
Q 9.1
Which of the following is a 3∘ amine?
(i) 1-methylcyclohexylamine (ii) Triethylamine
(iii) tert-butylamine (iv) N-methylaniline
Correct option: (ii) Triethylamine, (C2H5)3N.
Concept used. An amine's degree is decided by
how many carbon substituents sit on the nitrogen, not by
the carbon skeleton. 1∘ has one C-N bond, 2∘
has two, and 3∘ has three. The classification of the
carbon bearing -NH2 (e.g. tert-butyl) is irrelevant
for amine ordering.
(i) 1-methylcyclohexylamine: -NH2 on a ring carbon ⇒
only one C-N bond ⇒1∘.
(ii) Triethylamine N(C2H5)3: three ethyl groups on N⇒3∘.
(iii) tert-butylamine (CH3)3C-NH2: still only one
C-N bond ⇒1∘ (the carbon is 3∘, not the amine).
(iv) N-methylaniline C6H5-NH-CH3: two C-N bonds ⇒2∘.
Only triethylamine has three carbons on N; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Count-the-carbons angle. The cleanest discrimination here
is a one-step inspection: cover everything except the nitrogen atom
and count carbons bonded directly to N. The labels
``methyl'', ``cyclohexyl'' or ``tert-butyl'' on neighbouring
carbons are noise –- only C-N bonds set the amine class.
Run the count on each option. (i) 1-methylcyclohexylamine: a single
ring carbon carries NH2, so N touches one carbon
⇒ 1∘. (ii) Triethylamine N(C2H5)3: N
holds three ethyl groups, three C-N bonds ⇒ 3∘.
(iii) tert-butylamine (CH3)3C-NH2: the carbon is
3∘ but the nitrogen still sees only one C ⇒ 1∘.
(iv) N-methylaniline C6H5-NHCH3: two carbons on N⇒ 2∘. Only triethylamine clears the three-carbon
bar.
Triethylamine; option (ii).
Q 9.2
The correct IUPAC name for CH2=CHCH2 NHCH3 is 1.4cm.
(i) Allylmethylamine (ii) 2-amino-4-pentene
(iii) 4-aminopent-1-ene (iv) N-methylprop-2-en-1-amine
Correct option: (iv) N-methylprop-2-en-1-amine.
Concept used. For an amine, the IUPAC name is built from
the longest carbon chain bearing -NH- (or -NH2),
numbered so that the amine carbon takes the lowest locant. Any
group on N is indicated by an italic ``N-'' prefix. Double
bonds get their own locant (``-en-'').
Parent chain: the three-carbon CH2=CH-CH2- piece (the
N is attached to C-1, the =CH- to C-2).
Suffix: ``-amine'' on C-1 ⇒ ``prop-2-en-1-amine''.
Substituent on N: -CH3⇒ prefix ``N-methyl''.
Full name: N-methylprop-2-en-1-amine. The common name ``allylmethylamine'' is non-IUPAC.
N-methylprop-2-en-1-amine; option (iv).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Build-the-name angle. Draw the connectivity first: a
three-carbon chain CH2=CH-CH2- with NH(CH3) on its
terminal carbon. The parent must be the longest chain that
contains the amine carbon, which is the three-carbon ``propene''
chain. The amine is on C-1 and the double bond is between C-2 and
C-3 when numbered from the amine end, so the parent name is
prop-2-en-1-amine. The methyl group hanging off the nitrogen is
denoted with the locant ``N-'', giving the full name
N-methylprop-2-en-1-amine.
Option (iv).
Q 9.3
Amongst the following, the strongest base in aqueous medium is 1.4cm.
(i) CH3NH2 (ii) NCCH2NH2 (iii) (CH3)2NH (iv) C6H5NHCH3
Correct option: (iii)(CH3)2NH, dimethylamine.
Concept used. In aqueous medium the strongest base
combines electron-donating alkyl groups (raise lone-pair
availability) with good hydration of the conjugate
ammonium ion. 2∘ aliphatic amines balance these factors
best, giving the well-known order 2∘ > 1∘ ≈ 3∘ > NH3 ≫ aryl amines.
(i) CH3NH2: one +I methyl ⇒ moderately basic.
(ii) NCCH2NH2: -C#N is strongly -I⇒ withdraws density off N⇒ weak base.
(iii) (CH3)2NH: two +I methyls + two N-H
bonds still solvate the ammonium ion well ⇒ strongest.
(iv) C6H5NHCH3: lone pair on N delocalises
into the ring ⇒ aryl amine, weakest of the four.
(CH3)2NH is the strongest aqueous base; option (iii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Eliminate-the-bad angle. Two of the four options eliminate
themselves on sight. Option (ii) carries an -C#N group: cyano
is one of the most strongly -I substituents and pulls density
straight off the nitrogen lone pair ⇒ very weak base.
Option (iv) is an aromatic amine: the nitrogen lone pair conjugates
into the benzene ring, leaving little density for H+
acceptance –- weaker than even ammonia.
Between (i) and (iii) the deciding factors are inductive donation
and aqueous solvation. Dimethylamine has two +I methyl groups
plus two residual N-H bonds that the conjugate
(CH3)2NH2+ uses for hydrogen-bond solvation. That
combination beats methylamine (one methyl, three solvating
N-H) on inductive contribution and beats trimethylamine on
solvation. Hence dimethylamine sits at the top of the aqueous
basicity ladder among these four.
Option (iii)(CH3)2NH.
Q 9.4
Which of the following is the weakest Br"onsted base?
(i) Aniline (C6H5-NH2) (ii) Piperidine (cyclic 2∘ amine C5H10NH)
(iii) Cyclohexylamine (iv) CH3NH2
Correct option: (i) Aniline.
Concept used. Brnsted basicity of an amine is set by
how available the N lone pair is for protonation. Aromatic
amines like aniline drain the lone pair into the ring by resonance
⇒ much weaker base than alkylamines (where the lone
pair is fully localised on N and is reinforced by +I).
Aniline: lone pair on N overlaps with the benzene
ring; four resonance structures put N+ on the ring
⇒ lone pair is partly delocalised, basicity low (pKb ≈ 9.4).
Cyclohexylamine (pKb ≈ 3.4): strong +I from cyclohexyl, strong base.
Methylamine (pKb ≈ 3.4): one +I methyl, moderately strong.
Aniline is the weakest Br"onsted base among the four; option (i).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Sort-by-class angle. Group the four amines into
``aromatic'' (lone pair conjugated with a ring) versus
``aliphatic'' (lone pair localised on N). Only aniline sits
in the aromatic bucket; piperidine, cyclohexylamine and
methylamine are all aliphatic. The aromatic bucket loses about
five pKb units of basicity to lone-pair delocalisation
into the ring, so aniline (pKb ≈ 9.4) is
already an order of magnitude or more weaker than any of the
three aliphatic amines on this list. The answer is therefore
aniline without needing finer discrimination among the
aliphatic three.
Option (i) aniline.
Q 9.5
Benzylamine may be alkylated as
C6H5CH2NH2 + R-X -> C6H5CH2NHR.
Which of the following alkyl halides is best suited for this reaction through SN1 mechanism?
(i) CH3Br (ii) C6H5Br (iii) C6H5CH2Br (iv) C2H5Br
Correct option: (iii)C6H5CH2Br (benzyl bromide).
Concept used.SN1 proceeds through a
carbocation. The reaction rate scales with the stability
of that cation: resonance-stabilised cations (benzyl,
allyl) and 3∘ > 2∘ alkyl cations win; aryl
halides cannot do SN1 because phenyl cations are
extremely unstable.
(i) CH3-X and (iv) C2H5-X: primary alkyl halides
⇒ cation is CH3+ or C2H5+, both highly
unstable ⇒SN1 does not run; SN2 pathway only.
(ii) C6H5-Br: aryl halide; the C-Br bond is
sp2, SN1 would require a phenyl cation ⇒ impossible.
(iii) C6H5CH2-Br: ionises to the resonance-stabilised
benzylic cation C6H5-CH2+⇒ excellent SN1 substrate.
Benzyl bromide; option (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Cation-stability angle.SN1 rate is set by
how fast the alkyl halide ionises to a carbocation, which in turn
is set by how stable that cation is. Methyl and ethyl bromide
would have to give CH3+ and CH3CH2+ –- both
primary cations of unusable stability, ruling out SN1.
Bromobenzene cannot ionise at all because a phenyl cation
C6H5+ has no stabilising hyperconjugation and a vacant
orbital on sp2 carbon. Benzyl bromide, in contrast, ionises to
the benzylic cation that spreads its positive charge over the
ring via four canonical resonance structures, mimicking a
tertiary cation in stability. So benzyl bromide is the only
SN1-friendly halide in the list.
Option (iii)C6H5CH2Br.
Q 9.6
Which of the following reagents would not be a good choice for reducing an aryl nitro compound to an amine?
(i) H2 (excess)/Pt (ii) LiAlH4 in ether
(iii) Fe and HCl (iv) Sn and HCl
Correct option: (ii)LiAlH4 in ether.
Concept used. Aryl nitro compounds Ar-NO2 are
reduced to aryl amines Ar-NH2 by dissolving-metal
reductants in acidic medium (Sn/HCl, Fe/HCl), or by catalytic
hydrogenation (H2/Pt, Pd, Ni). LiAlH4 is unsuitable:
with aromatic nitro groups it tends to stop at azo
(Ar-N=N-Ar) or hydrazo (Ar-NH-NH-Ar)
compounds rather than going cleanly to Ar-NH2.
LiAlH4 delivers H- but does not cleanly cleave the second N-O bond on an aryl nitro group; product mixtures (azo/hydrazo) result.
LiAlH4 is the poor choice; option (ii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Pattern-recognition angle. The CBSE/NCERT textbook lists
exactly three ``approved'' routes from Ar-NO2 to Ar-NH2:
acidic dissolving-metal (Sn/HCl or Fe/HCl), and catalytic H2
over Pt/Pd/Ni. Anything outside that triad is the odd one out, and
here that role is filled by LiAlH4 in ether.
The chemical reason is that aromatic nitro groups are
π-conjugated with the ring, so simple hydride delivery stalls
at intermediate stages –- azoxy, azo and hydrazo species
(coloured by-products). Acidic conditions provide protons that
keep funnelling the intermediates onward; catalytic hydrogen
provides clean surface reduction. LiAlH4 provides neither
a proton bath nor a metal surface, so the reduction tarballs.
Option (ii)LiAlH4/ether is the wrong reagent.
Q 9.7
In order to prepare a 1∘ amine from an alkyl halide with simultaneous addition of one CH2 group in the carbon chain, the reagent used as source of nitrogen is 1.4cm.
(i) Sodium amide, NaNH2 (ii) Sodium azide, NaN3
(iii) Potassium cyanide, KCN (iv) Potassium phthalimide, C6H4(CO)2N-K+
Correct option: (iii) Potassium cyanide (KCN).
Concept used. To add one carbon while installing a
1∘ amine, the standard route is R-X KCN
R-CN LiAlH4 R-CH2-NH2. The nitrile carbon becomes the
new -CH2- in the amine. The other reagents give amines but
with no chain extension.
R-X + KCN -> R-CN + KX (SN2).
R-CN + 4 [H] LiAlH4 R-CH2-NH2 (one C added as the new -CH2- next to N).
NaNH2 would just deprotonate or give nitrene; NaN3 gives R-N3 -> R-NH2 with no chain extension; phthalimide (Gabriel) gives R-NH2 also with no chain extension.
KCN; option (iii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Match-the-constraint angle. The question demands two
things simultaneously –- the product is a primary amine, and the
carbon chain must grow by one CH2. Only the nitrile route
does both: KCN first delivers -CN onto R-X
via SN2, then catalytic or hydride reduction (Pt/Pd or
LiAlH4) of R-CN adds two hydrogens at carbon and
two at nitrogen to give R-CH2-NH2. The freshly installed
CH2 comes from the nitrile carbon, so the chain is one
carbon longer than the parent halide. The other three reagents
either keep the chain length the same (azide, phthalimide) or
fail to give a primary amine at all (sodium amide).
KCN; option (iii).
Q 9.8
The source of nitrogen in Gabriel synthesis of amines is 1.4cm.
(i) Sodium azide, NaN3 (ii) Sodium nitrite, NaNO2
(iii) Potassium cyanide, KCN (iv) Potassium phthalimide, C6H4(CO)2N-K+
Correct option: (iv) Potassium phthalimide.
Concept used.Gabriel synthesis converts a
1∘ alkyl halide into a pure 1∘ amine without
over-alkylation. The nitrogen source is potassium phthalimide,
made by reacting phthalimide with KOH. Its anion Phth-N-
does SN2 on R-X; subsequent acid or base
hydrolysis releases R-NH2 plus phthalic acid.
Deprotonation of phthalimide with KOH:
Phth-NH + KOH -> Phth-N- K+ + H2O
Hydrolysis with aqueous acid:
Phth-N-R + H3O+ -> R-NH2 + phthalic acid
(hydrazine NH2NH2 gives the same amine plus phthalhydrazide as the by-product).
Nitrogen comes from potassium phthalimide; option (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Definition-first angle. ``Gabriel'' literally names the
phthalimide route to primary amines, so the only acceptable
nitrogen donor is potassium phthalimide. The other three reagents
are the headline N-sources for sibling syntheses –- sodium azide
is the azide synthesis (gives R-N3, then R-NH2 after
reduction), NaNO2 is the diazotisation reagent and KCN is
the nitrile route (gives R-CN, then R-CH2-NH2 on
LiAlH4 reduction –- the chain grows by one carbon).
Potassium phthalimide; option (iv).
Q 9.9
Amongst the given set of reactants, the most appropriate for preparing 2∘ amine is 1.4cm.
(i) 2∘R-Br + NH3 (ii) 2∘R-Br + NaCN followed by H2/Pt
(iii) 1∘R-NH2 + RCHO followed by H2/Pt
(iv) 1∘R-Br (2 mol) + potassium phthalimide followed by H3O+/heat
Correct option: (iii) reductive amination of a 1∘ amine with an aldehyde.
Concept used.Reductive amination is the cleanest
laboratory route to 2∘ amines: a 1∘ amine condenses
with an aldehyde to give an imine, and the imine is reduced
catalytically to the 2∘ amine. No over-alkylation is
possible because the carbonyl supplies exactly one carbon
fragment.
R-NH2 + R2-CHO -> R-N=CH-R2 + H2O (imine; R2 is a generic second alkyl group).
R-N=CH-R2 + H2 Pt R-NH-CH2-R2 (2∘ amine).
(i) gives a mixture: R2NH, R3N, R4N+ side products.
(ii) gives R-CH2-NH2, a 1∘ amine (not 2∘).
(iv) Gabriel gives only 1∘ amines (single alkylation on phthalimide).
Reductive amination of a 1∘ amine with an aldehyde; option (iii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Stop-at-secondary angle. The constraint is to make
exactly a 2∘ amine, not a mixture. Direct alkylation
with NH3 overshoots into tertiary and quaternary
ammonium salts, and Gabriel is locked at primary by design.
The cyanide-plus-hydrogenation route gives a 1∘ amine
(R-CH2-NH2), not secondary. That leaves reductive
amination: a 1∘ amine and an aldehyde condense to a
single imine, hydrogenation reduces it cleanly to the
secondary amine, and the stoichiometry naturally caps the
process at one N-C bond addition. That single-step,
single-product profile is exactly what ``most appropriate''
demands.
Option (iii).
Q 9.10
The best reagent for converting 2-phenyl-propan-amide into 2-phenyl-propan-amine is 1.4cm.
(i) excess H2 (ii) Br2 in aqueous NaOH
(iii) iodine in the presence of red phosphorus (iv) LiAlH4 in ether
Correct option: (iv)LiAlH4 in ether.
Concept used. Reduction of an amide R-CONH2 with
LiAlH4 converts the carbonyl C=O into CH2 and
preserves the C-N bond without loss of carbon. So
the carbon count is unchanged –- 2-phenylpropanamide
(C6H5-CH(CH3)-CONH2, 9 C) → 2-phenylpropanamine
(C6H5-CH(CH3)-CH2-NH2, 9 C).
R-CONH2 + 4 [H] LiAlH4 R-CH2-NH2 + H2O.
(ii) Br2/NaOH is Hoffmann bromamide ⇒ would lose one C, giving 1-phenylethanamine (asked in Q11).
(i) Plain H2 does not reduce amides; (iii) is a P/I combination for halide reduction, not amides.
LiAlH4/ether; option (iv).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Keep-the-carbon angle. The two products in Q10 and Q11 are
constitutional cousins of the same amide, differing only by one
carbon. To go from R-CONH2 to R-CH2-NH2 (one more
carbon than the Hoffmann product), the C=O has to be reduced
all the way to CH2 while preserving the C-N bond –-
exactly what lithium aluminium hydride does to amides. So
LiAlH4 is the reagent that converts 2-phenylpropanamide
into 2-phenylpropanamine without losing a carbon.
Option (iv).
Q 9.11
The best reagent for converting 2-phenyl-propan-amide into 1-phenyl-ethan-amine is 1.4cm.
(i) excess H2/Pt (ii) NaOH/Br2
(iii) NaBH4/methanol (iv) LiAlH4/ether
Concept used.Hoffmann bromamide converts
R-CONH2 into R-NH2, dropping the carbonyl carbon as
Na2CO3. Apply to 2-phenylpropanamide C6H5-CH(CH3)-CONH2
(9 C) and the product is C6H5-CH(CH3)-NH2 (8 C) =
1-phenylethanamine.
Mechanism: N-bromination → deprotonation →α-elimination to isocyanate R-N=C=O→ hydrolysis to R-NH2.
(iv) LiAlH4 gives 2-phenylpropanamine (same C count), not 1-phenylethanamine.
Hoffmann bromamide with NaOH/Br2; option (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Lose-one-carbon angle. The product 1-phenylethanamine
C6H5-CH(CH3)-NH2 has one fewer carbon than the starting
2-phenylpropanamide C6H5-CH(CH3)-CONH2, so the route must
shed a carbon. Hoffmann bromamide degradation is the textbook
``one carbon shorter'' reaction –- treatment with Br2/NaOH
converts the primary amide to the corresponding amine while
expelling the carbonyl carbon as carbonate. So Br2/NaOH is
the right pick.
Option (ii).
Q 9.12
Hoffmann Bromamide Degradation reaction is shown by 1.4cm.
(i) ArNH2 (ii) ArCONH2 (iii) ArNO2 (iv) ArCH2NH2
Correct option: (ii)ArCONH2, a primary aryl amide.
Concept used. The Hoffmann bromamide rearrangement
requires a substrate with the -CONH2 functional group –-
i.e. a primary amide. The two N–H bonds are essential (one is
brominated, the other is deprotonated). Amines, nitro arenes and
-CH2NH2 are not amides and do not undergo this rearrangement.
Step 2: deprotonation, α-elimination to acyl nitrene; alkyl/aryl group migrates from C to N.
Step 3: the isocyanate R-N=C=O hydrolyses to R-NH2 + CO2.
N-substituted amides (R-CONH-R2) lack the second N–H and do not undergo Hoffmann.
Only primary amide ArCONH2 undergoes Hoffmann; option (ii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Functional-group-first angle. Hoffmann bromamide is named
after its substrate –- the primary bromamide, i.e. an
R-CONH2 amide that has had one N-H replaced by
N-Br. The rearrangement only fires on a primary amide that
carries two N-H bonds, one of which is brominated and the
other of which is deprotonated to drive the migration. Aryl
amines and aryl nitro compounds lack the carbonyl, while
ArCH2NH2 is again an amine, not an amide. Only
ArCONH2 qualifies.
Option (ii).
Q 9.13
The correct increasing order of basic strength for the following compounds is 1.4cm.
(I) Aniline (II) p-nitroaniline (III) p-toluidine
(i) II < III < I (ii) III < I < II (iii) III < II < I (iv) II < I < III
Correct option: (iv) II < I < III.
Concept used. Aromatic amine basicity is governed by how
much electron density sits on N. Electron-donating
para substituents (-CH3, +I, +H) push density onto N⇒ stronger base. Electron-withdrawing groups
(-NO2, -M, -I) pull density out of N via the ring
⇒ weaker base.
p-nitroaniline (II): -NO2 strongly withdraws by -M/-I⇒ weakest.
Aniline (I): only the ring delocalisation drains N⇒ intermediate.
p-toluidine (III): -CH3 donates by +I/hyperconjugation ⇒ strongest.
II < I < III; option (iv).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Substituent-effect angle. For substituted anilines, basicity
tracks the electron density on the N lone pair, which in turn
tracks what the para substituent does to the ring. An
electron-donating group like para-methyl pushes density into
the ring through +I (and weak hyperconjugation) and pulls it back
out toward N, boosting basicity. An electron-withdrawing
group like para-nitro does the opposite: it has -M and
-I, so it siphons density off N through resonance
structures that place a positive charge directly on the amino group
(-N+=C), crashing the basicity by several pKb
units. Aniline itself sits in the middle as the unsubstituted
benchmark. So the basicity order, weakest → strongest, is
p-NO2-C6H4-NH2 < C6H5-NH2 < p-CH3-C6H4-NH2.
II < I < III; option (iv).
Q 9.14
Methylamine reacts with HNO2 to form 1.4cm.
(i) CH3-O-N=O (ii) CH3-O-CH3 (iii) CH3OH (iv) CH3CHO
Correct option: (iii)CH3OH (methanol).
Concept used.1∘ aliphatic amines and HNO2
give an unstable aliphatic diazonium ion that fragments at once
to N2 plus a carbocation; water traps the cation to give
an alcohol. For methylamine the cation is CH3+ and the
isolated organic product is methanol.
CH3NH2 + HNO2 -> CH3-N2+ + 2 H2O.
CH3-N2+ -> CH3+ + N2 (the gas).
CH3+ + H2O -> CH3OH + H+.
Methanol, CH3OH; option (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Diazonium-then-water angle.1∘ aliphatic amines
plus HNO2 go through three quick steps: (1) protonation and
nitrosation of N to give an unstable diazonium ion
R-N#N+, (2) spontaneous loss of N2 (a fantastic
leaving group) to give a carbocation, (3) capture of the cation by
water to give the alcohol. For methylamine the cation is methyl,
and the trapped product is methanol. Brisk frothing of N2
is the gas, the alcohol is the liquid.
Option (iii)CH3OH.
Q 9.15
The gas evolved when methylamine reacts with nitrous acid is 1.4cm.
(i) NH3 (ii) N2 (iii) H2 (iv) C2H6
Correct option: (ii)N2.
Concept used. Primary aliphatic amines react with
HNO2 (generated in situ from NaNO2 + HCl) to
give an unstable aliphatic diazonium salt
R-N2+ X-. Unlike aryl diazonium salts (which are stable
at 0-5 ∘C), the aliphatic one decomposes
at once to release N2 and a carbocation that is
trapped by water to give the corresponding alcohol.
CH3NH2 + HNO2 -> CH3-N2+ + 2 H2O (diazotisation).
Spontaneous: CH3-N2+ -> CH3+ + N2 (the gas evolved).
Brisk evolution of N2 gas confirms a 1∘ aliphatic amine; option (ii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mechanism-shortcut angle. The diazonium cation
R-N#N+ is intrinsically unstable when R is an
aliphatic group, because the leaving fragment N2 is one of
the best leaving groups known and there is no aromatic ring to
delocalise the carbocation that forms. The instant the methyl
diazonium CH3-N2+ is generated, it spits out N2
quantitatively and gives a methyl cation that water grabs to give
methanol. The brisk frothing of N2 at room temperature is
the classroom test for a 1∘ aliphatic amine.
N2 gas evolves; option (ii).
Q 9.16
In the nitration of benzene using a mixture of conc. H2SO4 and conc. HNO3, the species which initiates the reaction is 1.4cm.
(i) NO2 (ii) NO+ (iii) NO2+ (iv) NO2-
Correct option: (iii) the nitronium ion NO2+.
Concept used. In the mixed-acid nitration of arenes,
H2SO4 protonates HNO3 and water leaves to generate
the nitronium ionNO2+. NO2+ is the
actual electrophile that attacks the benzene ring.
HNO3 + H2SO4 -> H2NO3+ + HSO4-.
H2NO3+ -> NO2+ + H2O.
C6H6 + NO2+ -> [arenium] -> C6H5-NO2 + H+.
NO, NO+ and NO2- never enter the mechanism in this medium.
NO2+; option (iii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Generate-the-electrophile angle. The mixed-acid nitrating
reagent is not nitric acid directly; the sulphuric acid (the
stronger acid) protonates nitric acid and ejects water to give
the linear nitronium ion O=N+=O. That cation has a
genuine vacant orbital on N and a +1 formal charge,
making it electrophilic enough to attack benzene's π
cloud. The neutral NO2 radical and the anion NO2-
are not electrophiles, while NO+ is the diazotisation
electrophile (different reaction).
NO2+.
Q 9.17
Reduction of aromatic nitro compounds using Fe and HCl gives 1.4cm.
(i) aromatic oxime (ii) aromatic hydrocarbon
(iii) aromatic primary amine (iv) aromatic amide
Correct option: (iii) aromatic primary amine.
Concept used.Fe/HCl is a dissolving-metal acidic
reductant that converts Ar-NO2 all the way to the
1∘ aryl amine Ar-NH2. The same outcome as Sn/HCl
or Zn/HCl, but cheaper ⇒ used industrially.
Overall: C6H5NO2 + 3 Fe + 7 HCl -> C6H5NH3+ Cl- + 3 FeCl2 + 2 H2O, then base gives C6H5NH2.
FeCl2 hydrolyses (FeCl2 + 2 H2O -> Fe(OH)2 + 2 HCl), regenerating HCl ⇒ only catalytic acid is needed.
1∘ aryl amine; option (iii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Reagent-recall angle. The trio Sn/HCl, Fe/HCl and
Zn/HCl all do the same job on aryl nitro compounds –- they
deliver electrons in an acidic medium and walk the nitro group
through nitroso and hydroxylamine to a clean primary aryl amine.
Fe/HCl is the cheapest variant and the only one used at industrial
scale (e.g. the Bechamp process for aniline). The other options
(oxime, hydrocarbon, amide) require entirely different reagents.
Aromatic primary amine; option (iii).
Q 9.18
The most reactive amine towards dilute hydrochloric acid is 1.4cm.
(i) CH3-NH2 (ii) (CH3)2NH (dimethylamine)
(iii) (CH3)3N (trimethylamine) (iv) Aniline (C6H5-NH2)
Correct option: (ii) dimethylamine, (CH3)2NH.
Concept used. Reactivity towards dilute HCl tracks
aqueous basicity. The well-known order is 2∘ > 1∘
≈ 3∘ > NH3 ≫ aryl amines. Dimethylamine
(CH3)2NH sits at the top of that ladder in the methyl
series.
(CH3)2NH: two +I methyls + one N-H for solvating H2N(CH3)2+⇒ strongest.
CH3NH2: one +I methyl + two N-H⇒ moderate.
(CH3)3N: three +I methyls but zero N-H⇒ poor cation solvation ⇒ weaker in water.
C6H5NH2: lone pair delocalised into ring ⇒ weakest.
(CH3)2NH is most reactive towards dilute HCl; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Compromise-wins angle. Three effects compete when an
amine encounters water. +I donation by alkyl groups grows from
1∘ → 3∘. Hydration of the ammonium cation
RnNH(4-n)+ improves as the number of N-H bonds
that can hydrogen-bond to water grows –- so it falls from
1∘ to 3∘. Steric crowding around N also
grows from 1∘ to 3∘, blocking protonation.
Dimethylamine maximises the sum: two methyls give plenty of +I,
one residual N-H still solvates the cation, and steric
crowding is moderate. Trimethylamine has more +I but loses to
poor cation solvation; methylamine has more solvation but loses
to weaker +I; aniline loses on every count because of
ring conjugation.
Option (ii) dimethylamine.
Q 9.19
Acid anhydrides on reaction with primary amines give 1.4cm.
(i) amide (ii) imide (iii) secondary amine (iv) imine
Correct option: (i) an amide.
Concept used.Acylation of amines: the amine
nitrogen attacks a carbonyl carbon of the acid anhydride,
displacing carboxylate, to give an N-acyl amide. With a
1∘ amine and acetic anhydride, the product is an
N-substituted acetamide.
Pyridine is often added to mop up the carboxylic acid that is co-produced.
Amide (R-NH-CO-R2); option (i).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Acyl-transfer angle. An acid anhydride has two acyl
groups linked through a shared oxygen and is a good acyl-donor
electrophile. A primary amine attacks one of the two carbonyl
carbons with its nitrogen lone pair, the tetrahedral
intermediate collapses with loss of carboxylate, and the product
is the N-acyl amide R-NH-CO-R2 plus a carboxylic acid
byproduct. The nitrogen now bears an acyl group rather than an
extra alkyl group, so the new C–N bond defines an amide, not a
secondary amine.
An amide; option (i).
Q 9.20
The reaction (with Cu metal in HCl) Ar-N2+ Cl- + Cu -> Ar-Cl + N2 + CuCl is named as 1.4cm.
(i) Sandmeyer reaction (ii) Gatterman reaction
(iii) Claisen reaction (iv) Carbylamine reaction
Correct option: (ii) Gatterman reaction.
Concept used. Two closely related transformations convert
an aryl diazonium chloride into an aryl halide. Sandmeyer
uses cuprous halide CuX in HX; Gatterman
uses freshly precipitated copper powder in HX. The
question shows plain Cu metal (not CuCl) above the arrow and
emits CuCl as a product –- the textbook signature of the
Gatterman variant.
Sandmeyer (with cuprous chloride in HCl):
ArN2+ Cl- + CuCl -> Ar-Cl + N2 + CuCl
(Cu(I) shuttles an electron back and forth.)
Gatterman (with Cu metal in HCl; CuCl is generated in situ):
ArN2+ Cl- + Cu -> Ar-Cl + N2 + CuCl
The equation in the question matches the Gatterman pattern exactly.
Gatterman reaction; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Read-the-arrow angle. The reagent over the arrow is plain
elemental copper in HCl, not the cuprous chloride that the
Sandmeyer reaction calls for. That single difference is decisive –-
Gatterman's modification was designed to dodge the awkward
preparation of CuCl by using freshly reduced copper powder
in concentrated HCl. The cuprous chloride that turns up on the
right-hand side of the given equation is generated in situ from
that copper, confirming the route. Both routes serve the same
synthetic end (replacing N2+ by Cl on the ring), but
the question's stoichiometry is uniquely Gatterman.
Gatterman reaction; option (ii).
Q 9.21
Best method for preparing primary amines from alkyl halides without changing the number of carbon atoms in the chain is 1.4cm.
(i) Hoffmann Bromamide reaction (ii) Gabriel phthalimide synthesis
(iii) Sandmeyer reaction (iv) Reaction with NH3
Correct option: (ii) Gabriel phthalimide synthesis.
Concept used. The question demands two things: (a) start
from alkyl halide, (b) keep the same carbon count.
Gabriel synthesis substitutes N from phthalimide onto
R-X via SN2 and then hydrolyses to R-NH2
–- the carbon skeleton from R-X is conserved exactly.
Reject (i) Hoffmann: starts from amide (R-CONH2), not R-X, and loses one C.
Reject (iii) Sandmeyer: starts from aryl diazonium, not alkyl halide.
(iv) NH3 + R-X works on alkyl halides but over-alkylates, giving a mix –- not the ``best'' method.
(ii) Gabriel: alkyl halide → pure 1∘ amine, same C count, no over-alkylation.
Gabriel phthalimide synthesis; option (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Constraints-first angle. Treat the question as a two-filter
sieve. Filter 1 –- the substrate is an alkyl halide
R-X: that immediately disqualifies Sandmeyer, which begins
from an aryl diazonium salt ArN2+ and never sees an alkyl
halide. Filter 2 –- the carbon count must not change: that
kicks out the Hoffmann bromamide route, which (a) starts from an
amide rather than R-X and (b) loses one carbon as
Na2CO3 during the rearrangement.
That leaves NH3-ammonolysis versus Gabriel synthesis. Plain
ammonia attacks R-X but the resulting amine is more
nucleophilic than ammonia itself, so a second, third and fourth
alkylation give a mess of 1∘/2∘/3∘/4∘
products that are tough to separate. Gabriel sidesteps this trap by
using phthalimide nitrogen, which can be alkylated only once, then
hydrolysing to release a pure 1∘ amine with the same
carbon skeleton as the parent halide.
Gabriel phthalimide synthesis; option (ii).
Q 9.22
Which of the following compounds will not undergo azo coupling reaction with benzene diazonium chloride?
(i) Aniline (ii) Phenol (iii) Anisole (iv) Nitrobenzene
Correct option: (iv) Nitrobenzene.
Concept used.Azo coupling is an
electrophilic aromatic substitution in which ArN2+ is a
weak electrophile and so needs a strongly activated
aromatic ring (e.g. -NH2, -OH, -OR). The ring
in nitrobenzene is strongly deactivated by -NO2
(-M, -I) and refuses coupling.
Aniline (-NH2): strong activator ⇒ couples at p to give p-aminoazobenzene (yellow dye).
Phenol (-OH): strong activator ⇒ couples at p to give p-hydroxyazobenzene (orange dye).
Anisole (-OMe): activator (lone pair on O donates) ⇒ couples at p.
Nitrobenzene (-NO2): strongly deactivated ⇒ no coupling.
Nitrobenzene fails to couple; option (iv).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Activator-vs-deactivator angle. Azo coupling needs an
electron-rich ring because the aryl diazonium cation is a feeble
electrophile. Aniline, phenol and anisole all carry strong
o/p-directing activators (-NH2, -OH, -OMe)
whose lone pairs flood the para carbon with electron density, so
each couples readily to give a coloured azo dye. Nitrobenzene
carries the strongly m-directing deactivator -NO2, which
makes the ring electron-poor at every coupling position. The
encounter complex with ArN2+ is simply not stable enough
to proceed, so nitrobenzene does not undergo azo coupling.
Option (iv).
Q 9.23
Which of the following compounds is the weakest Br"onsted base?
(i) Aniline (C6H5NH2) (ii) Cyclohexylamine (C6H11NH2)
(iii) Phenol (C6H5OH) (iv) Cyclohexanol (C6H11OH)
Correct option: (iii) Phenol.
Concept used. Basicity depends on (a) which heteroatom
holds the lone pair (N less electronegative than O
⇒ N bases stronger), and (b) whether the lone
pair is delocalised by an aromatic ring (-M via resonance
⇒ basicity drops sharply). Phenol combines both
penalties –- oxygen and ring conjugation –- making it the
weakest base in the set.
Cyclohexylamine: aliphatic amine, no resonance, lone pair on N⇒ strongest (pKb ≈ 3.4).
Cyclohexanol: aliphatic alcohol, O holds lone pair tightly ⇒ weaker base than amines.
Phenol: -OH on benzene ⇒ lone pair delocalised into ring; phenol is actually a weak acid (pKa ≈ 10), not a base ⇒ weakest base.
Phenol is the weakest Br"onsted base; option (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Heteroatom-and-ring angle. Run two filters across the
four candidates. Filter one: nitrogen bases beat oxygen bases of
the same skeleton, because nitrogen's lower electronegativity
lets it donate its lone pair more readily. That immediately
makes the two alcohols weaker bases than the two amines. Filter
two: aromatic substrates lose more basicity than aliphatic ones,
because the ring lone-pair conjugation pulls density away from
the heteroatom. So among the two alcohols, phenol is weaker than
cyclohexanol –- and phenol is actually acidic, not basic, on a
pKa scale. The weakest base of the four is therefore
phenol.
Option (iii).
Q 9.24
Among the following amines, the strongest Br"onsted base is 1.4cm.
(i) Aniline (C6H5-NH2) (ii) NH3
(iii) Pyrrole (aromatic 5-ring N–H) (iv) Pyrrolidine (saturated 5-ring N–H)
Correct option: (iv) Pyrrolidine.
Concept used. Pyrrolidine is a saturated 2∘
aliphatic amine ⇒ lone pair on N is fully
sp3 and available for protonation. Pyrrole is aromatic;
its N lone pair sits in the aromatic π system and is
not available for H+ acceptance. Aniline and NH3
are weaker than aliphatic amines.
Pyrrolidine: pKb ≈ 2.9 –- a strong base (like piperidine).
Pyrrole: pKb ≫ 14; using its N lone pair for H+ would destroy aromaticity ⇒ extremely weak base.
Pyrrolidine, option (iv), is by far the strongest base in the set.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Where-is-the-lone-pair angle. The cleanest discriminator
in this question is whether the nitrogen lone pair sits in the
aromatic π system or in a localised sp3 orbital. Pyrrole's
N donates its lone pair into the ring to complete the
6π aromatic count, so protonation of N would
destroy aromaticity –- a steep energy penalty that suppresses
its basicity to almost zero. Aniline's lone pair is partially
delocalised into the benzene ring, which weakens but does not
eliminate basicity. Ammonia is the unsubstituted benchmark.
Pyrrolidine is the only fully saturated, 2∘ aliphatic
amine in the set –- its lone pair is fully localised on
sp3 nitrogen and reinforced by two +I alkyl groups, so it
is the strongest base.
Option (iv) pyrrolidine.
Q 9.25
The correct decreasing order of basic strength of the following species is 1.4cm. H2O, NH3, OH-, NH2-
(i) NH2- > OH- > NH3 > H2O (ii) OH- > NH2- > H2O > NH3
(iii) NH3 > H2O > NH2- > OH- (iv) H2O > NH3 > OH- > NH2-
Correct option: (i)NH2- > OH- > NH3 > H2O.
Concept used. Two trends operate together: (a) anionic
forms (NH2-, OH-) are far stronger bases than the
neutral parents (NH3, H2O), because the lone pair is
unshared; (b) for the same charge type, nitrogen bases beat oxygen
bases (N less electronegative ⇒ donates lone
pair more readily).
NH2-: anion on N⇒ strongest base.
OH-: anion on O⇒ strong, but less so than NH2- (O holds lone pair more tightly).
NH3: neutral N⇒ moderate base (pKb ≈ 4.7).
H2O: neutral O⇒ weak base.
NH2- > OH- > NH3 > H2O; option (i).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Charge-then-electronegativity angle. Two independent
factors decide the ordering. The first is overall charge: a
formal negative on the heteroatom (as in NH2- or
OH-) makes the lone pair vastly more eager to grab
H+ than the neutral parents NH3 or H2O. The
second is which heteroatom carries the charge: for both the
charged and the neutral pair, the nitrogen species is a stronger
base because nitrogen is less electronegative and holds its lone
pair more loosely. Stacking the two filters gives
NH2- > OH- > NH3 > H2O.
Option (i).
Q 9.26
Which of the following should be most volatile?
(I) CH3CH2CH2NH2 (II) (CH3)3N
(III) CH3CH2-NH-CH3 (IV) CH3CH2CH3
Choose: (i) II (ii) IV (iii) I (iv) III
Correct option: (ii) IV, propane.
Concept used.Volatility is the inverse of
boiling point. Boiling point rises with the strength of
intermolecular attractions –- the dominant one here is
hydrogen bonding, which requires N-H or
O-H. Propane has only weak London forces; trimethylamine
has no N-H; methylethylamine has one N-H;
n-propylamine has two N-H. So the BP ladder is propane
< trimethylamine < methylethylamine <n-propylamine
⇒ propane is the most volatile.
Propane: BP -42 ∘C (no H-bonding).
Trimethylamine (3∘ amine): BP +3 ∘C (no N-H).
N-methylethylamine (2∘): BP ∼ 37 ∘C (one N-H).
n-Propylamine (1∘): BP 48 ∘C (two N-H).
Propane (IV) is the most volatile; option (ii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Count-the-NH angle. The four molecules have nearly the
same molar mass, so dispersion forces are roughly equal. The
volatility ordering then collapses to the number of N-H
bonds available for intermolecular hydrogen bonding. Propane has
none and only dispersion forces, so it has the lowest boiling
point and the highest volatility. Trimethylamine has no N-H
either, but its N lone pair can still accept hydrogen
bonds from neighbours (slight increase). N-methylethylamine adds
one N-H donor, n-propylamine adds two. So the volatility
ordering is propane > trimethylamine > N-methylethylamine
>n-propylamine, and the most volatile is propane.
Option (ii) IV (propane).
Q 9.27
Which of the following methods of preparation of amines will give same number of carbon atoms in the chain of amines as in the reactant?
(i) Reaction of nitrile with LiAlH4.
(ii) Reaction of amide with LiAlH4 followed by treatment with water.
(iii) Heating alkyl halide with potassium salt of phthalimide followed by hydrolysis.
(iv) Treatment of amide with bromine in aqueous solution of sodium hydroxide.
Correct option: (iii) Gabriel phthalimide synthesis.
Concept used. Audit each route for carbon-count change:
(i) Nitrile reduction: R-C#N + 4 [H] -> R-CH2-NH2 –- the nitrile C becomes the new CH2 in the amine. Carbon count rises by 1 relative to the parent halide; relative to the nitrile, same. Depends on how you count ``reactant''.
(ii) Amide reduction by LiAlH4: R-CONH2 -> R-CH2-NH2 –- same carbon count as the amide.
(iii) Gabriel: Phth-NK + R-X -> R-NH2 –- same carbon count as the alkyl halide (the only new C comes from phthalimide, which is removed on hydrolysis).
NCERT key explicitly marks (iii) as the correct answer (Gabriel preserves the carbon count of the starting alkyl halide exactly).
Gabriel synthesis preserves the carbon count of the alkyl halide ⇒ option (iii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Carbon-balance angle. The question asks which route maps
the reactant carbon framework one-to-one onto the amine. Gabriel
synthesis answers this cleanly when ``reactant'' is read as the
alkyl halide R-X –- the phthalimide nitrogen is grafted on
during alkylation, then sliced off during hydrolysis, so the
amine's carbon skeleton matches the halide's exactly. Hoffmann
bromamide instead loses one carbon as carbonate; nitrile
reduction gains one carbon from the nitrile C. Amide
reduction also preserves the count but the NCERT key flags only
the Gabriel route here.
Option (iii).
II. Multiple Choice Questions (Type-II)
Q 9.28
Which of the following cannot be prepared by Sandmeyer's reaction?
(i) Chlorobenzene (ii) Bromobenzene (iii) Iodobenzene (iv) Fluorobenzene
Correct options: (iii) and (iv) –- iodobenzene and fluorobenzene.
Concept used. Sandmeyer's reaction needs the cuprous
halide CuX (X = Cl, Br) to deliver X to the aryl
radical generated from ArN2+. Cuprous iodide is not
required: I- from KI reacts directly with the
diazonium salt –- so iodobenzene is made without Cu (not strictly
``Sandmeyer''). Cuprous fluoride doesn't work either; aryl
fluorides use the Balz–Schiemann route via ArN2+ BF4-.
Cl, Br: classic Sandmeyer with CuCl/HCl or CuBr/HBr.
I: needs only KI, no Cu; not a true Sandmeyer.
F: Sandmeyer fails; use HBF4 then heat (Balz–Schiemann).
Sandmeyer fails for Ar-I and Ar-F –- options (iii), (iv).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Halide-by-halide angle. Walk down the four answer choices
and ask, for each, whether the textbook Sandmeyer recipe (cuprous
halide in HX on ArN2+) actually applies. Chlorobenzene and
bromobenzene fall straight into Sandmeyer's sweet spot –- Cu(I)
chloride and Cu(I) bromide are the classic salts that mediate
ArN2+ -> Ar-Cl and ArN2+ -> Ar-Br via a single-electron-transfer mechanism.
Iodobenzene is the trap. It is made from ArN2+, but no
copper is required: iodide is such a good nucleophile that a plain
solution of KI at room temperature does the job, so the
transformation is not Sandmeyer in the strict sense. Fluorobenzene
is even further outside Sandmeyer's reach –- cuprous fluoride is
unstable, so the route used industrially is Balz–Schiemann: trap
the diazonium as the tetrafluoroborate ArN2+ BF4- and heat
the dry salt. Hence the two ``not by Sandmeyer'' aryl halides are
iodide and fluoride.
Options (iii) and (iv).
Q 9.29
Reduction of nitrobenzene by which of the following reagents gives aniline?
(i) Sn/HCl (ii) Fe/HCl (iii) H2-Pd (iv) Sn/NH4OH
Correct options: (i), (ii) and (iii) –- Sn/HCl, Fe/HCl, and H2/Pd.
Concept used. Reducing C6H5-NO2 to C6H5-NH2
needs acidic dissolving-metal conditions (Sn/HCl, Fe/HCl,
Zn/HCl) or catalytic hydrogenation (H2/Pt, Pd, Ni). Under
basic/neutral conditions (Sn/NH4OH, LiAlH4),
reduction halts at intermediate species (azoxybenzene, azobenzene,
hydrazobenzene), not aniline.
C6H5NO2 + 3 Sn + 7 HCl -> C6H5NH3+ Cl- + 3 SnCl2 + 2 H2O, then base gives C6H5NH2.
Fe/HCl is industrial: cheap, FeCl2 hydrolyses to regenerate HCl (catalytic acid).
Sn/NH4OH is basic ⇒ stops at azoxy/azo stage, not aniline.
(i), (ii), (iii) all give aniline; (iv) does not.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Acidic-vs-basic angle. The textbook rule is that
nitrobenzene reduces cleanly to aniline only when the reducing
system supplies both electrons and protons. The acidic
dissolving-metal trio –- Sn/HCl, Fe/HCl, Zn/HCl –- meets both
requirements: the metal donates electrons, the strong acid keeps
the intermediates protonated and funnels them downhill all the way
to Ar-NH2. Catalytic hydrogen over Pd is the other approved
route; the metal surface dissociates H2 into atomic
hydrogen that walks the nitro group through nitroso, hydroxylamine
and finally amine.
Sn in NH4OH flips this picture upside down. The medium is
basic, so the protonation steps that drive the reduction past
azoxy- and azo-benzene never happen. Reduction simply stalls at
azoxy- or azo-benzene (coloured by-products), and aniline is not
formed. That is the standard trick question on this topic.
(i), (ii), (iii) all give aniline; (iv) stops short.
Q 9.30
Which of the following species are involved in the carbylamine test?
(i) R-NC (ii) CHCl3 (iii) COCl2 (iv) NaNO2 + HCl
Correct options: (i) and (ii) –- R-NC and CHCl3.
Concept used. The carbylamine test for 1∘
amines uses CHCl3 and alcoholic KOH. The dichlorocarbene
CCl2 generated in situ reacts with R-NH2 to
give the foul-smelling alkyl isocyanide (carbylamine)
R-NC. Phosgene COCl2 and NaNO2/HCl are reagents of
different reactions (Schotten-Baumann and diazotisation,
respectively).
CHCl3 + KOH -> CCl2 + KCl + H2O (α-elimination).
R-NH2 + CCl2 + 2 KOH -> R-NC + 2 KCl + 2 H2O.
R-NC is the product whose obnoxious smell flags a 1∘ amine.
Species involved: CHCl3 and R-NC; options (i), (ii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Mechanism-checklist angle. List what enters and what
leaves the carbylamine test. Inputs: a primary amine
R-NH2, chloroform CHCl3, and alcoholic potassium
hydroxide. Output: alkyl isocyanide R-NC (the source of
the obnoxious smell) plus KCl and water. Of the four candidate
species in the question, two appear in the balanced reaction:
chloroform on the reactant side, alkyl isocyanide on the product
side. Phosgene and the diazotising mixture NaNO2/HCl are
reagents for other named transformations (Schotten-Baumann,
diazotisation) and never enter the carbylamine equation.
Options (i) and (ii).
Q 9.31
The reagents that can be used to convert benzene diazonium chloride to benzene are 1.4cm.
(i) SnCl2/HCl (ii) CH3CH2OH (iii) H3PO2 (iv) LiAlH4
Correct options: (ii) and (iii) –- ethanol and hypophosphorous acid.
Concept used. Replacing the -N2+ group of an aryl
diazonium salt by an -H atom (i.e. deamination) is
done with mild reducing agents that supply H. or H-.
The two NCERT-listed reagents are ethanol
(CH3CH2OH, which is oxidised to acetaldehyde) and
hypophosphorous acid (H3PO2).
SnCl2/HCl reduces Ar-N2+ to Ar-NH-NH2 (phenylhydrazine), not benzene. LiAlH4 is not the textbook reagent for this replacement.
Use CH3CH2OH or H3PO2; options (ii), (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Replace-by-H angle. Removing the diazonium group as
N2 and dropping a hydrogen in its place requires a mild
hydride or hydrogen-atom donor. The NCERT-approved reagents are
ethanol and hypophosphorous acid: ethanol is oxidised to
acetaldehyde while delivering H. to the aryl radical, and
H3PO2 is oxidised to H3PO3 while doing the same.
SnCl2/HCl over-reduces the diazonium to a
phenylhydrazine Ar-NH-NH2 (Bechamp variant), and
LiAlH4 tends to give phenylhydrazine or hydrazo products
rather than clean replacement by hydrogen. So the right pair is
ethanol and hypophosphorous acid.
Options (ii) and (iii).
Q 9.32
The product of the following reaction is 1.4cm.
Acetanilide + Br2/CH3COOH ⟶ ?
(i) p-bromoacetanilide (ii) o-bromoacetanilide
(iii) m-bromoacetanilide (iv) 2,4,6-tribromoacetanilide
Correct options: (i) and (ii) –- p-bromoacetanilide (major) and o-bromoacetanilide (minor).
Concept used. Acetanilide C6H5-NHCOCH3 has the
-NHCOCH3 group, a mild activator (lone pair on N
delocalised into both the carbonyl and the ring), and an
o/p-director with a strong steric bias for p.
Br2/CH3COOH at room temperature gives mono-bromination
at the activated o/p positions; over-bromination (as in aqueous
Br2) does not occur in acetic acid solvent.
-NHCOCH3 activates the ring less strongly than -NH2 does (amide resonance into C=O steals some lone-pair density from the ring).
Bromination occurs only once ⇒ mono-substituted product.
Para is preferred over ortho on steric grounds (the bulky NHCOCH3 group blocks ortho approach).
Mainly p-bromoacetanilide with some o-bromoacetanilide; options (i), (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Tamed-activator angle. Acetylation of aniline replaces a
free amine -NH2 (very strong activator) by an amide
-NHCOCH3 (mild activator), because amide resonance siphons
some of the nitrogen lone pair into the C=O rather than
into the ring. Under those conditions a single electrophilic
bromination at the activated o/p positions is the natural
outcome. The para position dominates because the bulky
NHCOCH3 blocks ortho approach. Hence the products are
p-bromoacetanilide (major) and o-bromoacetanilide (minor); the
tribromo product does not form under these mild conditions.
Options (i) and (ii).
Q 9.33
Arenium ion involved in the bromination of aniline is 1.4cm.
(i) Cyclohexadienyl-NH2+-H-Br at C-2 (o-attack arenium)
(ii) Cyclohexadienyl with NH2, with + at C-5 and H, Br at C-2 (o-attack second resonance form)
(iii) Para arenium: =NH2+ at C-1, H, Br at C-4
(iv) Ortho arenium: NH2 at C-1, + at C-3, H, Br at C-2
Correct options: (i), (ii) and (iii) –- all three are valid arenium-ion (Wheland) intermediates for the bromination of aniline.
Concept used. An arenium ion (Wheland complex)
forms when the electrophile Br+ adds to a ring carbon,
generating a cyclohexadienyl cation. For an o/p-director like
-NH2, the + charge can be delocalised onto the carbon
bearing -NH2 via a resonance structure of type
R2C=NH2+, giving an extra-stable iminium contributor at
ortho and para positions only (not meta).
Ortho attack: Br goes to C-2; the + charge delocalises through three positions, one of which puts it on the N atom as =NH2+ at C-1 (structure (i)). Other resonance forms place + on C-3 or C-5 (structure (ii)).
Para attack: Br goes to C-4; the + charge delocalises and one resonance form again puts it on N as =NH2+ at C-1 (structure (iii)).
Meta attack (option (iv)) does not enjoy the iminium stabilisation ⇒ not a favoured arenium and not the answer.
Bromination via o- and p-arenium intermediates; options (i), (ii), (iii).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Resonance-stabilisation angle. When Br+ attacks
aniline, the resulting Wheland intermediate has the positive
charge spread across three ring carbons. For o- and p-attack,
one of those carbons is the same carbon that holds the NH2
group –- meaning the nitrogen lone pair can hop in and form a
genuine C=N+ bond, producing an iminium resonance
structure that drops the energy of the intermediate
substantially. For m-attack, the positive carbons never line up
with the NH2-bearing carbon, so the iminium structure is
not accessible and the arenium is much less stable. That is why
all three arenium drawings labelled o and p are valid (they
are merely different resonance contributors of the same
intermediate) and the meta drawing is not.
Options (i), (ii) and (iii).
Q 9.34
Which of the following amines can be prepared by Gabriel synthesis?
(i) Isobutyl amine (ii) 2-Phenylethylamine
(iii) N-methylbenzylamine (iv) Aniline
Correct options: (i) and (ii) –- isobutyl amine and 2-phenylethylamine.
Concept used. Gabriel synthesis works only when the alkyl
halide can undergo SN2 on the phthalimide
anion. Two structural conditions: (a) the halide must be a
1∘ (or 2∘) alkyl halide, and (b) the product
must be a primary amine (R-NH2). Aryl halides
(C6H5-X) do not do SN2 and 2∘ amines
(like N-methylbenzylamine) cannot come from a single alkylation
of phthalimide.
(i) Isobutyl amine: from (CH3)2CH-CH2-Br (1∘ alkyl halide) ⇒ Gabriel OK.
(ii) 2-Phenylethylamine: from C6H5CH2CH2-Br (1∘ benzylic-adjacent) ⇒ Gabriel OK.
(iii) N-methylbenzylamine is 2∘ (C6H5CH2-NH-CH3); Gabriel gives only 1∘ amines ⇒ fails.
(iv) Aniline would need C6H5-Br + phthalimide anion ⇒SN2 on sp2 carbon fails.
Only (i) and (ii) are accessible by Gabriel synthesis.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Filter-the-targets angle. Gabriel synthesis succeeds only
when two boxes are ticked simultaneously: the starting halide must
be an alkyl halide that can undergo backside SN2 on the
phthalimide nitrogen, and the final amine must be primary (so that
one alkylation is enough). Run each target through this two-step
sieve.
Aniline (iv) needs C6H5-X; that is an aryl halide where the
sp2 carbon refuses SN2, so Gabriel is dead on
arrival. N-methylbenzylamine (iii) is a secondary amine
C6H5CH2-NH-CH3; phthalimide nitrogen carries only one
ionisable hydrogen, so it can only deliver one alkyl group and
hence only primary amines. Isobutyl amine (i) traces back to
isobutyl bromide (CH3)2CH-CH2-Br –- a clean primary alkyl
halide that does SN2 smoothly. 2-Phenylethylamine (ii)
traces back to C6H5-CH2-CH2-Br, also primary alkyl
(benzylic-adjacent, not benzylic itself) –- again Gabriel-friendly.
Hence only the latter two work.
(i) and (ii) only.
Q 9.35
Which of the following reactions are correct?
(i) (CH3)2CHCl + 2 NH3 -> (CH3)2CH-NH2 + NH4Cl
(ii) (CH3)2CHCl + aq. KOH -> propene (CH3-CH=CH2)
(iii) Cyclohexyl chloride + alc. KOH → cyclohexene
(iv) (CH3)2CH-NH2 + HNO2 0 ∘C (CH3)2CH-OH
Correct options: (i) and (iii).
Concept used. Audit each equation against the standard reactivity rules:
(i) Ammonolysis of 2∘ alkyl halide with NH3 gives a 1∘ amine ⇒ correct.
(ii) Aqueous KOH on a 2∘ alkyl halide gives the alcohol (substitution), not the alkene. Alkene formation needs alcoholic KOH (E2).
(iv) Isopropylamine + HNO2 gives isopropanol only at room temperature, not at 0 ∘C as written for aryl diazotisation; the equation pattern is right (aliphatic diazonium → alcohol + N2), but the temperature label of 0 ∘C is the giveaway for the aryl pattern. The NCERT key marks (iv) as incorrect because the equation includes the 0 ∘C condition that belongs to aryl diazotisation.
(i): (CH3)2CHCl + 2 NH3 -> (CH3)2CHNH2 + NH4Cl –- one mole of NH3 is the nucleophile, the other neutralises HCl. Correct.
(ii): aqueous KOH supplies OH-, the strong nucleophile in a polar medium. Substitution to give 2-propanol dominates; elimination is minor. The equation as written (forming the alkene) is wrong.
(iii): alcoholic KOH supplies OH- in a less-solvating medium where the small base is forced to deprotonate β-H, giving cyclohexene by E2.
(iv): isopropylamine (1∘ aliphatic) does give 2-propanol with HNO2, but it does so spontaneously at room temperature –- the 0 ∘C label is reserved for aryl amines.
Correct equations: (i) and (iii).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Equation-audit angle. Score each equation independently
on whether reagent, solvent, temperature and product match.
Equation (i) is the textbook ammonolysis of a secondary alkyl
halide –- a single SN2 delivers isopropylamine and an
NH4Cl byproduct, all consistent. Equation (iii) is the
textbook E2 elimination on cyclohexyl chloride with alcoholic
KOH, giving cyclohexene. Equation (ii) mismatches solvent and
product: aqueous KOH on a secondary alkyl halide gives the
alcohol via SN2, not the alkene that the equation
shows. Equation (iv) has the right product (isopropanol from a
primary aliphatic amine with HNO2) but the wrong
temperature label of 0 ∘C, which belongs to
aryl diazotisation; aliphatic diazonium ions decompose at room
temperature. So only (i) and (iii) are fully correct.
Options (i) and (iii).
Q 9.36
Under which of the following reaction conditions, aniline gives p-nitro derivative as the major product?
(i) Acetyl chloride/pyridine followed by reaction with conc. H2SO4 + conc. HNO3.
(ii) Acetic anhydride/pyridine followed by conc. H2SO4 + conc. HNO3.
(iii) Dil. HCl followed by reaction with conc. H2SO4 + conc. HNO3.
(iv) Reaction with conc. HNO3 + conc. H2SO4.
Correct options: (i) and (ii) –- acylation (either reagent) first, then nitration gives p-nitroaniline as the major product.
Concept used. Direct nitration of aniline gives mostly
the m-isomer because H2SO4 protonates -NH2 to the
m-directing -NH3+. Acetylating the amine first (with
CH3COCl or (CH3CO)2O in pyridine) converts
-NH2 to the much less basic -NHCOCH3, which stays
o/p-directing under the strong-acid nitration conditions and
gives mainly p.
(i) Acetyl chloride/pyridine on aniline → acetanilide. Nitration of acetanilide → p-nitroacetanilide (major). Hydrolysis (acid) gives p-nitroaniline.
(ii) Acetic anhydride/pyridine → acetanilide; same outcome as (i).
(iii) Dil. HCl protonates aniline to anilinium (m-directing) before nitration ⇒ m-nitroaniline, not p.
(iv) Direct nitration: same anilinium-induced m-directing ⇒ m-nitroaniline mainly.
Acylate N first (any acyl chloride or anhydride) then nitrate; options (i), (ii).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Protect-before-nitrate angle. Aniline cannot be nitrated
cleanly without protection because the nitrating mixture itself
protonates -NH2 to -NH3+, which is strongly
m-directing and also so deactivating that the ring is prone to
oxidation. The fix is to convert -NH2 into an
electron-balanced amide -NHCOCH3 before nitration. Acetic
anhydride or acetyl chloride, both in pyridine, do exactly that,
yielding acetanilide whose -NHCOCH3 is mildly activating
and o/p-directing with a strong para bias on steric grounds.
Nitration of acetanilide therefore gives mostly the p-nitro
product, and acid hydrolysis unmasks p-nitroaniline.
Options (i) and (ii).
Q 9.37
Which of the following reactions belong to electrophilic aromatic substitution?
(i) Bromination of acetanilide (ii) Coupling reaction of aryldiazonium salts
(iii) Diazotisation of aniline (iv) Acylation of aniline
Correct options: (i) and (ii) –- bromination of acetanilide and azo coupling.
Concept used.Electrophilic aromatic substitution
(EAS) replaces a ring H with an electrophile while retaining
aromaticity. The two-step arenium-ion mechanism applies. Reactions
at the nitrogen of an amine (diazotisation, acylation) are
not EAS even though aromatic amines are involved.
(i) Acetanilide + Br2/CH3COOH: Br+ attacks the ring at p⇒ EAS.
(ii) Coupling: ArN2+ is the electrophile attacking another activated aromatic ring (phenol, aniline) ⇒ EAS.
(iii) Diazotisation: reaction at -NH2 (not on ring); NO+ attacks N⇒ not EAS.
(iv) Acylation of aniline: (CH3CO)2O acylates N to give acetanilide ⇒ not EAS.
(i) and (ii) are EAS; (iii), (iv) react at N, not the ring.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Site-of-attack angle. Run a single test on every option:
does the electrophile bond to a ring carbon, with loss of
the ring proton? If yes, the reaction is electrophilic aromatic
substitution; otherwise it is something else even if an arene is
involved.
(i) Bromination of acetanilide –- Br+ generated from
Br2/CH3COOH attacks the para carbon of the strongly
activated ring; aromaticity is restored on loss of H+.
Classic EAS. (ii) Azo coupling –- the aryl diazonium cation
ArN2+ is itself a mild electrophile that attacks an
electron-rich arene (phenol or aniline) at the para ring carbon,
giving the azo dye and losing the para H+. Also EAS.
(iii) Diazotisation –- NO+ (from HNO2/HCl)
adds to the nitrogen lone pair of ArNH2, never
touching the ring carbons. (iv) Acylation of aniline –- acetic
anhydride acylates the nitrogen of -NH2 to give
acetanilide, again sparing the ring. So only the first two count.
(i) and (ii).
III. Short Answer Type
Q 9.38
What is the role of HNO3 in the nitrating mixture used for nitration of benzene?
Answer:HNO3 acts as a base in the
nitrating mixture: it accepts H+ from H2SO4 and
then loses water to generate the actual electrophile, the
nitronium ionNO2+.
Concept used. Generating an electrophile strong enough
to attack benzene requires the cooperation of two acids:
H2SO4 is the stronger acid (proton donor) and HNO3
is the weaker acid; in their mutual presence, HNO3
behaves as a base.
Overall: HNO3 provides the nitrogen by being protonated to give nitronium.
HNO3 acts as a base, supplying NO2+ (the nitronium electrophile) on protonation by H2SO4.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Generate-the-electrophile angle. The whole point of the
mixed-acid recipe is to take an unreactive aromatic ring and a
mildly electrophilic HNO3 and combine them into a
genuinely electrophilic attack on the arene. To do that, the
nitric acid has to give up its -OH as water and leave a
nitrogen bearing a vacant orbital. Sulphuric acid, being the
stronger acid, protonates nitric acid; the protonated nitric
acid then dehydrates to the linear O=N+=O ion. So
HNO3's role is twofold –- it is the source of the
NO2 group and it temporarily becomes a base by accepting
the sulphuric acid proton.
HNO3 behaves as a base; the product NO2+ is the active electrophile.
Q 9.39
Why is the -NH2 group of aniline acetylated before carrying out nitration?
Answer: Acetylation converts the strongly activating but
basic -NH2 into the mildly activating, non-basic
-NHCOCH3. This prevents (a) protonation of -NH2 by
H2SO4 to the m-directing -NH3+, and (b)
oxidation of aniline by HNO3.
Concept used. Three problems arise if aniline is
nitrated directly:
H2SO4 protonates the basic -NH2 to -NH3+, which is strongly m-directing ⇒ mostly m-nitroaniline.
HNO3 is a strong oxidant; aniline is electron-rich ⇒ partial oxidation gives tarry residues.
Even if some unprotonated aniline reacts, over-nitration to 2,4,6- and beyond is possible.
Acetyl protection keeps N neutral, prevents oxidation, and steers nitration cleanly to the para position.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Two-pronged angle. Direct nitration of aniline fails for
two parallel reasons. The sulphuric acid in the nitrating mix
protonates the basic -NH2 to -NH3+, flipping the
director from o/p (the free amine) to m (the ammonium). The
nitric acid in the mix oxidises the very electron-rich ring,
giving black tarry oxidation products. Acetylation in pyridine
solves both problems at once. The amide -NHCOCH3 is so
non-basic that even H2SO4 cannot protonate it, so the
o/p-directing character is preserved through the nitration.
The amide is also a much weaker activator than the free amine,
so the ring is no longer prone to oxidation. After nitration,
acid hydrolysis of the acetamide gives back the free amine,
unmasked at the right ring position.
Acetylation protects -NH2 from protonation and oxidation, preserving o/p-directing nitration.
Q 9.40
What is the product when C6H5CH2NH2 reacts with HNO2?
Product: Benzyl alcohol, C6H5CH2OH.
Concept used. Benzylamine is a 1∘aliphatic
amine (the -NH2 sits on an sp3 benzylic carbon, not on
the ring), so it follows the aliphatic-amine + HNO2 pattern:
unstable diazonium ⇒N2 escapes ⇒H2O traps the carbocation ⇒ alcohol.
C6H5CH2NH2 + HNO2 -> C6H5CH2-N2+ + 2 H2O.
C6H5CH2-N2+ -> C6H5CH2+ + N2 (benzylic cation is resonance-stabilised, decomposition is fast).
C6H5CH2+ + H2O -> C6H5CH2OH + H+.
Benzyl alcohol, C6H5CH2OH, with brisk evolution of N2.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Classify-first angle. Decide whether the amine is
aromatic (lone pair attached directly to sp2 ring carbon) or
aliphatic (lone pair on an sp3 carbon). Benzylamine has the
-CH2- buffer between the ring and the nitrogen, so it is
aliphatic. Aliphatic primary amines plus HNO2 always
collapse via an unstable diazonium to the corresponding alcohol
plus N2 gas, so C6H5CH2NH2 gives C6H5CH2OH.
Benzyl alcohol C6H5CH2OH with N2 release.
Q 9.41
What is the best reagent to convert nitrile to primary amine?
Answer: Lithium aluminium hydride, LiAlH4 in dry
ether, or catalytic hydrogenation with sodium in alcohol (Mendius
reduction).
Concept used.Nitrile reduction adds two
H atoms across each of the two C-Nπ bonds,
converting R-C#N into R-CH2-NH2 (a 1∘
amine that has one more carbon than the parent halide).
Catalytic hydrogenation (H2/Ni, Pd, or Pt) also works but can over-reduce in some cases.
LiAlH4 in ether (or Na/C2H5OH) reduces nitriles cleanly to 1∘ amines.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Hydride-delivery angle. A nitrile's C#N triple
bond is reduced in two stages: first to an imine R-CH=NH,
then to the amine R-CH2-NH2. Lithium aluminium hydride is
strong enough to push through both steps in a single ether
solution, with no isolation of the imine intermediate, and gives
the primary amine cleanly. The Mendius alternative (Na in
ethanol) is the cheaper bench-scale option used in pre-hydride
laboratory practice. Either way, the product is the primary
amine R-CH2-NH2.
LiAlH4 in ether (or Na/C2H5OH).
Q 9.42
Give the structure of `A' in the following reaction.
4-methyl-2-nitroaniline (i) NaNO2 + HCl, 273-278 K(ii) H3PO2, H2O A.
(The substrate has CH3 at C-4, NO2 at C-2, NH2 at C-1 of benzene.)
Answer: `A' is 3-nitrotoluene (m-nitrotoluene),
CH3-C6H4-NO2 with NO2 at the meta position relative to CH3.
Concept used. Diazotisation at 0-5 ∘C
(273-278 K) converts Ar-NH2 to Ar-N2+;
subsequent reduction with H3PO2/H2O replaces -N2+
by -H (deamination). The starting material has CH3
at C-4 and NO2 at C-2 relative to the NH2
group at C-1. After deamination at C-1, only CH3 and
NO2 remain on the ring, CH3 at the original C-4
position and NO2 at the original C-2 –- but after dropping
NH2, the ring is renumbered so that the substituents now
end up meta to each other.
Diazotisation: Ar-NH2 + NaNO2 + 2 HCl -> Ar-N2+ Cl- + NaCl + 2 H2O at 273-278 K.
Ring map: C-1 was NH2, C-2 was NO2, C-4 was CH3. After removing NH2, the remaining substituents are NO2 and CH3 on positions that translate to 1,3 (meta).
Track-the-substituents angle. The substrate has three
ring substituents: NH2 at C-1, NO2 at C-2 and
CH3 at C-4 of benzene. Step (i) diazotises the amine to
-N2+ Cl- at C-1, holding it at the cold 0-5
∘C window that stabilises the aryl diazonium
salt. Step (ii) hands off a hydrogen atom from H3PO2 to
the aryl radical, releasing N2 and replacing -N2+
by -H. So NH2 at C-1 simply disappears. What is
left is a benzene ring with NO2 (originally at C-2) and
CH3 (originally at C-4) –- which sit meta to each
other in the renumbered product. The product is 3-nitrotoluene.
3-nitrotoluene (m-nitrotoluene).
Q 9.43
What is Hinsberg reagent?
Answer:Hinsberg reagent is benzenesulphonyl
chloride, C6H5-SO2Cl.
Concept used. The Hinsberg test uses
C6H5SO2Cl to distinguish 1∘, 2∘ and
3∘ amines. The reagent's S(=O)2 is a strong
electrophile; an amine's lone pair displaces Cl- to give
a sulphonamide.
1∘ amine: R-NH2 + C6H5SO2Cl -> C6H5SO2NHR. The N-H is acidic (sulphonyl is strongly EW) ⇒ soluble in alkali.
2∘ amine: R2NH + C6H5SO2Cl -> C6H5SO2NR2. No N-H left ⇒ insoluble in alkali.
3∘ amine: no N-H to lose ⇒ does not react.
Hinsberg reagent = C6H5SO2Cl (benzenesulphonyl chloride); used to distinguish 1∘/2∘/3∘ amines by alkali solubility.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Reagent-plus-pattern angle. Hinsberg's reagent is
benzenesulphonyl chloride, C6H5-SO2-Cl. The strongly
electrophilic sulphonyl sulphur is attacked by the amine lone pair,
displacing chloride and producing a sulphonamide whose properties
diagnose the amine class.
With a primary amine, the resulting C6H5-SO2-NHR retains an
N-H that is rendered acidic by the two electron-withdrawing
oxygens on sulphur, so NaOH deprotonates it and the salt
dissolves. With a secondary amine, the sulphonamide
C6H5-SO2-NR2 has no N-H left and stays as an
alkali-insoluble solid. A tertiary amine has no N-H at the
outset and does not even react. Comparing the three test tubes
fixes the class of an unknown amine on the bench.
Hinsberg reagent is C6H5SO2Cl.
Q 9.44
Why is benzene diazonium chloride not stored and is used immediately after its preparation?
Answer: Benzene diazonium chloride
C6H5-N2+ Cl- is thermally unstable above
5 ∘C. On warming, even gently, it decomposes
to give phenol, N2 and HCl (or chlorobenzene if dry); on
prolonged storage even in solution it slowly decomposes by the
same routes. So it must be prepared at 0-5 ∘C
and used immediately in the downstream reaction.
Concept used. The arenium-stabilised Ar-N#N+
cation is more stable than its aliphatic cousin (which dies on
contact with water), but at higher temperatures the loss of
N2 becomes spontaneous because N2 is an outstanding
leaving group.
In dry solid form, on standing: C6H5-N2+ Cl- -> C6H5-Cl + N2 (the dry salt can even detonate).
Thermal instability of Ar-N2+; prepared cold (0-5 ∘C) and used at once to avoid loss of N2 to give phenol or chlorobenzene.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Thermal-decomposition angle. A diazonium salt is a
delicate species; the bond linking the aryl ring to the
-N#N+ group breaks easily because N2 is one of
the most thermodynamically favourable leaving groups. In an
aromatic compound, ring resonance partially stabilises the
cation, but only at very low temperature. Warming above
∼ 5 ∘C supplies enough thermal energy to
eject N2, and in aqueous solution water immediately
traps the aryl cation as phenol. So the standard recipe is to
diazotise in an ice bath at 0-5 ∘C and
pipe the cold solution directly into the next reaction –- never
to bottle and store.
Diazonium chloride is unstable above 5 ∘C; preparation and use must be back-to-back at 0-5 ∘C.
Q 9.45
Why does acetylation of -NH2 group of aniline reduce its activating effect?
Answer: The N lone pair of aniline, originally
free to push into the ring (strong +M activator), is now
shared with the carbonyl O via the amide resonance
R-NH-CO-R2 <-> R-NH+=C(O-)-R2. The lone pair
becomes less available to the ring ⇒ ring activation
drops sharply.
Concept used. The amide carbonyl is a π-acceptor that
competes with the benzene ring for the nitrogen lone pair.
Resonance with C=O is energetically more favourable than
resonance with the benzene ring (carbonyl is more polarisable),
so most of the lone pair is locked into the N-C=O system.
In aniline: lone pair N -> ring (+M); ring activation ≈ 106× benzene.
In acetanilide: lone pair is shared between N -> ring and N -> C=O; the carbonyl wins the larger share.
Net: NHCOCH3 is still an activator (mild) but much weaker than NH2 (strong).
Amide resonance with C=O pulls the lone pair off the ring, dropping the activating power of -NHCOCH3 relative to -NH2.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Lone-pair-competition angle. In free aniline the
nitrogen lone pair is entirely available to push into the benzene
ring, which is why -NH2 is a powerful o/p-directing
activator. Acylation grafts a carbonyl onto the nitrogen, and
the same lone pair now has two competitors –- the benzene ring
and the carbonyl oxygen. The carbonyl wins the larger share
because amide resonance (N-C=O <-> N+=C-O-)
is highly stabilising; the ring loses most of its lone-pair
contribution and therefore most of its activation. Acetanilide
is still an activator but only mildly so, which is why
nitration of acetanilide can be controlled to give predominantly
the para product without ring oxidation.
Amide resonance steals the lone pair from the ring, lowering activation.
Q 9.46
Explain why MeNH2 is a stronger base than MeOH.
Concept used. Basicity (pKb) is measured by
how readily the heteroatom's lone pair grabs H+. The more
available the lone pair (low electronegativity, good
solvation of the resulting cation), the stronger the base.
Electronegativities: χ(N) = 3.04, χ(O) = 3.44.
Since O holds its lone pair more tightly than N, methylamine's N donates its lone pair to H+ more readily than methanol's O does.
Conjugate-acid view: CH3NH3+ (pKa ≈ 10.6) is much weaker than CH3OH2+ (pKa ≈ -2). Weaker conjugate acid ⇒ stronger base.
N is less electronegative than O, so its lone pair is more available for protonation; hence MeNH2 is the stronger base.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Electronegativity angle. The deciding factor for a
neutral heteroatom base is how tightly the atom holds its lone
pair. Electronegativity is the direct proxy. Nitrogen sits at
χ = 3.04 on the Pauling scale; oxygen at χ = 3.44.
Lower electronegativity means a more diffuse, more donatable
lone pair, so methylamine's nitrogen donates electrons to
H+ far more readily than methanol's oxygen does.
Cross-check with conjugate-acid strengths: CH3NH3+ has
pKa ≈ 10.6, while CH3OH2+ has
pKa ≈ -2. Methanol's conjugate acid is roughly
1012 times stronger, so methanol itself is ∼ 1012
times weaker as a base. The pattern generalises across the table
–- amines beat alcohols of the same skeleton in basicity by
many orders of magnitude.
N lone pair is more available than O lone pair, so MeNH2 ≫ MeOH as a base.
Q 9.47
What is the role of pyridine in the acylation reaction of amines?
Answer: Pyridine acts as a base that neutralises
the HCl (or CH3COOH) liberated during acylation,
keeping the amine deprotonated (and therefore nucleophilic) and
pushing the equilibrium towards the amide product. Pyridine also
catalyses the reaction via a nucleophilic-catalysis pathway
(N-acyl pyridinium intermediate).
Concept used. If the by-product acid (HCl from acyl
chloride or CH3COOH from anhydride) is allowed to
accumulate, it would protonate the amine to R-NH3+, which
has no lone pair available for further attack and so the
acylation stalls. Pyridine sops up that acid as
C5H5N · H+ X-.
Acylation: R-NH2 + R2-COCl -> R-NH-CO-R2 + HCl.
Pyridine traps HCl: HCl + C5H5N -> C5H5NH+ Cl-.
Without pyridine: HCl + R-NH2 -> R-NH3+ Cl-, the salt is unreactive.
Pyridine is the acid scavenger (and a mild nucleophilic catalyst); it keeps the amine free and pulls the acylation forward.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Acid-scavenger angle. Every acylation of an amine
produces an acidic by-product –- HCl when the acyl source is
an acid chloride, carboxylic acid when it is an anhydride. If
that acid is left in solution it protonates the amine
nucleophile to R-NH3+, which loses its lone pair and
stops attacking the carbonyl. Pyridine, with pKb
≈ 8.8, is basic enough to grab the proton from HCl but
not basic enough to compete with the amine for the acyl
electrophile; it also weakly catalyses the acylation through a
short-lived N-acyl-pyridinium intermediate. So pyridine keeps
the reaction running cleanly to the amide.
Pyridine removes the acid by-product, keeping the amine free.
Q 9.48
Under what reaction conditions (acidic/basic), the coupling reaction of aryldiazonium chloride with aniline is carried out?
Answer:Mildly acidic (pH ≈ 4-5) –-
or equivalently very weakly basic but not alkaline.
Concept used. Two species must coexist for the coupling
to work: (a) the aryl diazonium cation ArN2+ (stable
only below 5 ∘C and in mildly acidic medium;
strong base destroys it as diazoate Ar-N=N-O-), and
(b) the nucleophilic free amine ArNH2 (lost under strongly
acidic conditions because -NH2 is protonated to the
unreactive -NH3+).
In strong acid: ArNH2 + H+ -> ArNH3+, no lone pair, no coupling.
In strong base: ArN2+ + OH- -> Ar-N=N-OH -> Ar-N=N-O- (diazoate), no electrophile.
At pH ∼ 4-5 (mildly acidic / weakly basic), enough free aniline (lone pair) coexists with enough ArN2+ to react.
Mildly acidic conditions (pH 4–5); the medium keeps both partners alive for coupling.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Both-must-survive angle. The trick to picking the right
pH for azo coupling is to ask which form of each partner is
catalytically active and what range of pH lets both forms
coexist. For aniline coupling the diazonium needs a mildly
acidic medium (so it does not convert to the unreactive
diazoate), and the aniline nitrogen needs to keep its lone pair
(so the medium cannot be strongly acidic enough to protonate
the amine to ArNH3+). Both conditions are met at pH
≈ 4-5, which the NCERT calls ``mild basic / weak
acidic''. So coupling of aryldiazonium chloride with aniline is
done in mild acid.
Mildly acidic (∼ pH 4–5) conditions.
Q 9.49
Predict the product of reaction of aniline with bromine in a non-polar solvent such as CS2.
Product: A mixture of 2-bromoaniline and 4-bromoaniline (mainly p-bromoaniline).
Concept used. Aniline is strongly activating; in
aqueous bromine water it gives 2,4,6-tribromoaniline at once.
In a non-polar solvent (CS2) the -NH2 is not
protonated and the reaction is gentler –- bromination stops at
the monosubstituted stage, giving mainly p-bromoaniline plus
some o-bromoaniline.
Generate Br+: Br2 polarises on approach to the electron-rich ring.
EAS at o/p: -NH2 is an o/p-director by +M.
In CS2, only one Br installs ⇒ mono-bromination at o and p.
!%
[See diagram in the PDF version]
%
Mixture of 2-bromoaniline and 4-bromoaniline (p-product dominant).
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Solvent-control angle. The two canonical bromination
outcomes of aniline are dictated entirely by solvent. In water,
Br2 is heterolytically polarised by the highly solvating
medium, releasing Br+ that finds the aniline ring extremely
electron-rich at three positions (ortho, ortho ' and
para). All three positions react, giving 2,4,6-tribromoaniline
as a white precipitate –- the standard qualitative test for aniline.
Switching the solvent to CS2 removes the polarising medium.
Br+ is generated only sluggishly, and the lone pair on
-NH2 is not protonated, so the ring is still activated but
substitution slows down dramatically. The first bromine installs at
the activated o or p position, after which the ring is less
reactive and the second/third Br+ attack is suppressed. The
isolable products are 2-bromoaniline (minor) and 4-bromoaniline
(major), the latter favoured for steric reasons over the
ortho positions.
Arrange the following compounds in increasing order of dipole moment. CH3CH2CH3, CH3CH2NH2, CH3CH2OH
Order:CH3CH2CH3 < CH3CH2NH2 < CH3CH2OH.
Concept used. Dipole moment μ scales with the
electronegativity difference across the polar bond.
χ(C) = 2.55, χ(N) = 3.04, χ(O) = 3.44.
The C-O dipole (Δχ = 0.89) is much larger than the
C-N dipole (Δχ = 0.49); the C-C bond in
propane has Δχ = 0, so propane is essentially apolar.
Propane: only C-H and C-C bonds; μ ≈ 0.08 D.
Ethylamine: C-N and N-H polar bonds; μ ≈ 1.22 D.
Ethanol: C-O and O-H polar bonds; μ ≈ 1.69 D.
CH3CH2CH3 < CH3CH2NH2 < CH3CH2OH.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Electronegativity-difference angle. Molecular dipole
moment is the vector sum of bond dipoles, so it scales with the
electronegativity difference Δχ across each polar bond
and with the geometry that decides whether they add or cancel.
For the three molecules here the geometry is similar (terminal
heteroatom on an ethyl chain), so the per-bond dipole reads almost
directly into the molecular dipole.
Propane has only C-C and C-H bonds, both with
Δχ near zero, so its dipole moment is essentially zero
(∼ 0.08 D). Ethylamine introduces a C-N bond
(Δχ = 0.49) and two N-H bonds (Δχ = 0.84),
giving μ ≈ 1.22 D. Ethanol has a C-O
bond (Δχ = 0.89) and an O-H bond
(Δχ = 1.24), each more polar than its nitrogen
counterpart, giving μ ≈ 1.69 D. So the dipole
order matches the electronegativity-difference order exactly:
propane < ethylamine < ethanol.
C3H8 < C2H5NH2 < C2H5OH.
Q 9.51
What is the structure and IUPAC name of the compound, allyl amine?
Answer: Structure –- CH2=CH-CH2-NH2. IUPAC name
–- prop-2-en-1-amine.
Concept used. The common name ``allyl'' is the
CH2=CH-CH2- group; adding -NH2 gives allylamine. To
name it by IUPAC: longest chain that includes the amine carbon
is propene; amine on C-1, double bond between C-2 and C-3
⇒ prop-2-en-1-amine.
Functional priority: amine > alkene, so the chain is numbered from the amine end.
C-1: CH2-NH2; C-2 and C-3: CH=CH2.
Locants: ``2-en'' for the double bond, ``1-amine'' for -NH2⇒ prop-2-en-1-amine.
Allyl amine =CH2=CH-CH2-NH2= prop-2-en-1-amine.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Common-to-IUPAC angle. The trivial name allylamine
encodes the connectivity: an allyl group CH2=CH-CH2- on
nitrogen, giving the three-carbon primary amine
CH2=CH-CH2-NH2. To convert to IUPAC, pick the longest
chain that includes the amine carbon –- here, the three-carbon
propene chain –- and number it so the principal characteristic
group (the amine) gets the lower locant. The amine ends up on
C-1 and the double bond between C-2 and C-3, giving
prop-2-en-1-amine.
CH2=CH-CH2-NH2; prop-2-en-1-amine.
Q 9.52
Write down the IUPAC name of C6H5-N(CH3)2 (N,N-dimethyl benzene amine).
Answer:N,N-dimethylbenzenamine (also written
N,N-dimethylaniline).
Concept used. For an aryl amine, the IUPAC name uses
``benzenamine'' (or the retained name ``aniline'') as the parent,
with N-substituents prefixed by italic ``N-''. The compound
C6H5-N(CH3)2 has two methyl groups on nitrogen ⇒
``N,N-dimethylbenzenamine''.
Parent: benzene with -NH2 = benzenamine (or aniline).
N-substituents: two methyls on nitrogen ⇒ ``N,N-dimethyl''.
Full IUPAC name: N,N-dimethylbenzenamine; equivalent common name: N,N-dimethylaniline.
N,N-dimethylbenzenamine (= N,N-dimethylaniline).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
N-locant angle. An aniline derivative with all
substituents on the nitrogen (rather than on the ring) is named
by leaving the parent as benzenamine and prefixing the
substituents with the locant ``N''. Two methyl groups on the
nitrogen of aniline give N,N-dimethylbenzenamine (or, using the
retained name, N,N-dimethylaniline). The italicised N's are
required to distinguish nitrogen substitution from C-ring
substitution.
N,N-dimethylbenzenamine.
Q 9.53
A compound Z with molecular formula C3H9N reacts with C6H5SO2Cl to give a solid, insoluble in alkali. Identify Z.
Answer: Z is ethylmethylamine, CH3-NH-C2H5, a 2∘ amine.
Concept used. Behaviour with the Hinsberg reagent
(C6H5SO2Cl) is a fingerprint of amine class. (a) 1∘ amine
gives an alkali-soluble sulphonamide (acidic N-H). (b) 2∘
amine gives an alkali-insoluble sulphonamide (no N-H). (c) 3∘
amine does not react.
Molecular formula C3H9N has degree of unsaturation = 0, so Z is acyclic and saturated.
Possible structures: n-C3H7-NH2 (1∘), (CH3)2CH-NH2 (1∘), CH3-NH-C2H5 (2∘), (CH3)3N (3∘).
``Reacts with Hinsberg'' ⇒ not 3∘. ``Solid insoluble in alkali'' ⇒2∘ amine.
The only C3H9N2∘ amine is ethylmethylamine CH3-NH-C2H5.
Z is ethylmethylamine, CH3-NH-C2H5, which gives N-ethyl-N-methylbenzene sulphonamide (alkali-insoluble).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Class-then-isomer angle. Approach this as a two-stage
deduction. First, the Hinsberg behaviour pins the class: a
sulphonamide that is insoluble in alkali can only come from
a secondary amine, because primary amines yield alkali-soluble
sulphonamides (acidic N-H) and tertiary amines do not react
with Hinsberg's reagent at all.
Second, the molecular formula C3H9N has degree of
unsaturation zero, so the parent is acyclic and saturated. The
four possible isomers are n-propylamine, isopropylamine,
N-methylethylamine, and trimethylamine. The first two are
primary, trimethylamine is tertiary, leaving N-methylethylamine
CH3-NH-C2H5 as the only C3H9N secondary amine.
That isomer fits every clue: it reacts with PhSO2Cl to
give N-ethyl-N-methylbenzene sulphonamide, which has no acidic
N-H and so stays insoluble in alkali.
Z = ethylmethylamine CH3-NH-C2H5.
Q 9.54
A primary amine R-NH2 can be reacted with CH3-X to get a secondary amine R-NHCH3, but the only disadvantage is that 3∘ amine and quaternary ammonium salts are also obtained as side products. Suggest a method where R-NH2 forms only the 2∘ amine.
Method: Use the carbylamine reaction followed
by catalytic hydrogenation.
R-NH2 CHCl3 / KOH R-NC H2 / Pd R-NHCH3
Concept used. The carbylamine route gives the
2∘ amine R-NH-CH3 in two clean steps: (a)
formation of alkyl isocyanide R-NC from R-NH2 with
CHCl3/alc. KOH (the two N-H are replaced by a
single N=C bond); (b) catalytic hydrogenation of
R-NC over Pd reduces the N=C bond to N-CH2-
=N-CH3 (because the carbon already carries no other H), giving the
2∘ amine without any further alkylation possible.
Net: R-NH2 -> R-NH-CH3, exactly one methyl added on N, no 3∘ or 4∘ side products.
R-NH2 CHCl3/KOH R-NC H2/Pd R-NHCH3 stops cleanly at the 2∘ amine.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Self-stopping-route angle. The reason direct alkylation
of a primary amine cannot be stopped at the secondary stage is
that each newly-formed amine is more nucleophilic than the one
that came before, so methylation just keeps going. The
carbylamine workaround sidesteps this by hand-installing the
methyl as an isocyanide in two non-amine steps. First, chloroform
plus alcoholic potash converts R-NH2 to the foul-smelling
alkyl isocyanide R-NC (a C=N bond, not a
nucleophilic amine). Second, catalytic hydrogenation reduces
that triple bond to a single N-CH3, giving exactly the
2∘ amine and stopping there because the new amine has
no available carbon nucleophile to over-react with. Cleanly
single-substituted.
Carbylamine (CHCl3/KOH) then H2/Pd.
Q 9.55
Complete the following reaction:
Phenol ArN2+ Cl-OH- ?
Product:p-Hydroxyazobenzene (when Ar = C6H5), the orange azo dye HO-C6H4-N=N-C6H5.
Concept used.Azo coupling: in mild alkaline
medium, phenol is deprotonated to the highly activated phenoxide
ion C6H5O-, which attacks the weak electrophile
ArN2+ at its para position to give an azo
compound. The orange/yellow colour comes from the extended
π-conjugated Ar-N=N-Ar2 chromophore.
Deprotonation: C6H5OH + OH- -> C6H5O- + H2O.
EAS coupling: ArN2+ + C6H5O- -> p-HO-C6H4-N=N-Ar + H+.
For the generic aryl, the product is p-aryloxoazobenzene; for Ar = C6H5, p-hydroxyazobenzene.
0.9!%
[See diagram in the PDF version]
%
Product = p-hydroxyazobenzene, an orange azo dye, formed by para coupling of phenoxide with aryl diazonium cation.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Phenoxide-EAS angle. Azo coupling needs a strongly
electron-rich aromatic partner because the diazonium cation is a
weak electrophile. Phenol meets that bar barely; phenoxide
C6H5O- blows past it, with the formal negative charge on
oxygen flooding the ring with electron density at the o and
p positions. Mildly alkaline medium is therefore ideal: it
generates the phenoxide while still being mild enough not to
destroy the diazonium as the unreactive diazoate. The product is
the para-coupled azo compound HO-C6H4-N=N-Ar, with the
extended π system that gives azo dyes their characteristic
orange to red colour.
p-hydroxyazobenzene, the orange para azo dye.
Q 9.56
Why is aniline soluble in aqueous HCl?
Concept used. Although aniline itself is a sparingly
soluble oily liquid (the -NH2 lone pair is partly delocalised
into the ring), its conjugate acid anilinium chlorideC6H5NH3+ Cl- is a fully ionic salt –- it dissolves in
water just as ammonium chloride does.
C6H5NH2 + HCl -> C6H5NH3+ Cl- (proton transfer).
The anilinium cation is hydrated by water (H-bonds to the three N-H bonds and ion–dipole attractions).
Cl- is independently hydrated ⇒ the salt dissolves freely in aqueous HCl.
Aniline + HCl gives the water-soluble salt anilinium chloride, hence the apparent solubility in aqueous HCl.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Salt-formation angle. The trick to seeing why aniline
``dissolves'' in HCl is to separate the free amine from
its protonated form. Free aniline is a neutral, oily, weakly
polar molecule held in water mainly by a single N-H ⋯ O
contact; it spreads as droplets on the water surface and is only
sparingly soluble.
Add HCl and the picture changes. Aniline is still a
Br nsted base (a feeble one, pKb ≈ 9.4) but
HCl is a strong acid, so proton transfer is essentially
complete: C6H5NH2 + HCl -> C6H5NH3+ Cl-. The product is
an ionic salt –- a fully charged anilinium cation hydrated by
water (ion–dipole plus three N-H ⋯ O hydrogen bonds)
and a chloride anion that water hydrates independently. Ionic
salts dissolve readily in water, so the apparent dissolution of
aniline in aqueous HCl is really the dissolution of the
salt anilinium chloride.
Aniline forms water-soluble anilinium chloride with HCl.
Q 9.57
Suggest a route by which the following conversion can be accomplished: cyclohexanecarboxamide → N-methylcyclohexylamine (c-C6H11-NH-CH3).
Concept used. Two named reactions string together: (a) Hoffmann
bromamide chops the carbonyl off the amide and gives the
1∘ amine cyclohexylamine; (b) Carbylamine grafts a single
methyl-equivalent (-NC) onto N which is then
reduced to -NH-CH3. Cleaner than direct methylation, which
over-alkylates.
Two-named-reactions angle. Read the target backwards.
N-methylcyclohexylamine has a methyl on a cyclohexyl nitrogen, so
the immediate precursor is something that, on reduction, deposits
a single CH3 on the amine nitrogen –- the canonical
candidate is cyclohexylisocyanide c-C6H11-NC, whose
N=C bond reduces under H2/Pd to N-CH3. That
isocyanide in turn comes from cyclohexylamine via the carbylamine
reaction with CHCl3/KOH. Finally, cyclohexylamine comes
from cyclohexanecarboxamide via Hoffmann bromamide degradation,
which drops the carbonyl carbon. Three named-reaction steps,
clean conversion.
Identify A and B in the following reaction:
2-(2-chloroethyl)cyclohexan-1-one KCN A H2/Pd B.
Answers.
A: 2-(2-cyanoethyl)cyclohexan-1-one –- the
-CH2-CH2-Cl tail of the substrate has been converted to
-CH2-CH2-CN by SN2 displacement of Cl- by CN-.
B: 2-(3-aminopropyl)cyclohexan-1-one –- the
nitrile of A has been reduced to a primary amine
-CH2-CH2-CH2-NH2 (the chain has gained one CH2).
Concept used. Two single-step transformations: SN2 with KCN on a primary alkyl chloride gives a nitrile (chain +1 C); H2/Pd reduces the nitrile to a 1∘ amine (with no further change in carbon count).
The cyclohexanone C=O does not reduce under H2/Pd at room temperature with normal pressure ⇒ ketone survives.
A = 2-(2-cyanoethyl)cyclohexan-1-one; B = 2-(3-aminopropyl)cyclohexan-1-one.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Two-step-sequence angle. The first arrow over KCN
is the textbook substitution that swaps a chloride for a cyanide.
The -CH2-CH2-Cl tail is a primary alkyl chloride, so the
SN2 goes smoothly, and the cyclohexanone ring is not
touched. The product A still bears the ketone but now has a
-CH2-CH2-CN chain. The second arrow over H2/Pd
reduces the nitrile to a primary amine (-CN becomes
-CH2-NH2, adding one CH2 to the carbon count). The
ring carbonyl survives because the Pd-catalysed conditions are
mild enough not to touch a ketone. So B is the amino-ketone with
a -CH2-CH2-CH2-NH2 tail.
A = 2-(2-cyanoethyl)cyclohexan-1-one; B = 2-(3-aminopropyl)cyclohexan-1-one.
Q 9.59
How will you carry out the following conversions?
(i) Toluene →p-toluidine. (ii) p-toluidine diazonium chloride →p-toluic acid.
Part (i): Toluene →p-toluidine.
[label=()]
Nitration: C6H5CH3 HNO3 / H2SO4 p-O2N-C6H4-CH3 (with some o-isomer; separate by fractional crystallisation).
Reduction: p-O2N-C6H4-CH3 Fe / HCl p-H2N-C6H4-CH3 (p-toluidine).
Part (ii): p-toluidine diazonium chloride →p-toluic acid (p-CH3-C6H4-COOH).
Functional-interconversion angle. Part (i) is a
straightforward two-step: nitrate toluene with mixed acid to
install -NO2 at p (with o as minor, easily separated)
and then reduce with Fe/HCl (cheaper than Sn/HCl) to give
p-toluidine. Part (ii) is a Sandmeyer-then-hydrolysis pair:
replacing -N2+ by -CN uses cuprous cyanide
CuCN in KCN; the resulting nitrile is then hydrolysed by
hot aqueous acid all the way to the carboxylic acid, giving
p-toluic acid. Each step is a standard textbook entry; the
overall sequence builds a -COOH at the same ring position
where the original -NH2 sat.
See answer above.
Q 9.60
Write following conversions:
(i) nitrobenzene → acetanilide; (ii) acetanilide →p-nitroaniline.
Part (i): Nitrobenzene → acetanilide.
[label=()]
Reduce nitrobenzene to aniline: C6H5-NO2 Sn / HCl C6H5-NH2.
Acylate aniline with acetic anhydride in pyridine: C6H5-NH2 + (CH3CO)2O pyridine C6H5-NHCOCH3 + CH3COOH.
Part (ii): Acetanilide →p-nitroaniline.
[label=()]
Nitrate with mixed acid: C6H5-NHCOCH3 HNO3 / H2SO4 p-O2N-C6H4-NHCOCH3.
Concept used. Part (i) reduces nitrobenzene to aniline
and acetylates. Part (ii) exploits acetanilide's o/p-directing
-NHCOCH3 to control nitration to the para position, then
hydrolyses off the acetyl group.
Sequences shown above. The protecting acetyl group is the trick that makes Part (ii) selective.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Protecting-group angle. Part (i) is a two-step
preparation of acetanilide: reduce nitrobenzene to aniline with
Sn/HCl (acidic dissolving metal), then acetylate the
amine with acetic anhydride in pyridine (the pyridine soaks up
the acetic acid by-product). Part (ii) needs p-nitroaniline,
which cannot be made cleanly by direct nitration of aniline
because the strong acid medium protonates the amine. The fix is
to start from acetanilide (where the amine is already
protected), nitrate with mixed acid to give predominantly the
para-nitro product, and finally hydrolyse off the acetyl group
to unmask p-nitroaniline. The acetyl protecting group is the
unsung hero of the sequence.
See sequences above.
Q 9.61
A solution contains 1 g mol each of p-toluene diazonium chloride and p-nitrophenyl diazonium chloride. To this, 1 g mol of alkaline solution of phenol is added. Predict the major product. Explain your answer.
Major product: the azo dye coupling p-nitrophenyl
diazonium with phenol, i.e. 4-nitro-4'-hydroxyazobenzenep-O2N-C6H4-N=N-C6H4-OH-p, not the p-toluene
version.
Concept used.Azo coupling is electrophilic
aromatic substitution; rate scales with the electrophilicity of
the diazonium cation. -NO2 at the para position of the
diazonium is strongly -M/-I and withdraws electron
density, making the diazonium more electrophilic. -CH3 in
the toluene-diazonium is mildly +I and weakly donates,
lowering the electrophilicity. So the nitro-substituted
diazonium wins the race.
Phenol in alkali → phenoxide (the nucleophile).
Two competing electrophiles in solution: p-O2N-C6H4-N2+ (more electrophilic) and p-CH3-C6H4-N2+ (less electrophilic).
Phenoxide couples preferentially with the stronger electrophile ⇒p-nitrophenyldiazonium wins.
Phenol preferentially couples with p-nitrophenyldiazonium chloride because -NO2 raises the electrophilicity of the diazonium cation; product is 4-hydroxy-4'-nitroazobenzene.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Electrophilicity-decides-the-race angle. Two aryl
diazonium cations compete for one mole of phenoxide. Coupling is
electrophilic aromatic substitution, so the diazonium that is
more electrophilic wins. The p-nitrophenyldiazonium cation
carries a strongly -M/-I nitro group para to the
-N2+, which sucks electron density out of the
-N=N- unit and concentrates positive charge there –-
making it a much hotter electrophile. The p-toluenediazonium
has a mildly +I methyl that donates a little density and
softens the electrophile. So phenoxide couples preferentially
with the nitro diazonium, giving 4-hydroxy-4'-nitroazobenzene
as the major product.
4-Hydroxy-4'-nitroazobenzene (orange-red).
Q 9.62
How will you bring out the conversion: p-nitro-aniline → 3,4,5-tribromo-nitro-benzene?
Strategy. Use Br2/CH3COOH to brominate the
ring of p-nitroaniline (the activating NH2 directs Br
to its two ortho positions, here both meta to the para-NO2),
then diazotise and remove the amine via H3PO2 to leave
the three bromines on the ring.
[label=()]
Bromination at both ortho positions to NH2:
p-O2N-C6H4-NH2 + 2 Br2 CH3COOH 2,6-dibromo-4-nitroaniline
(the bromines sit at C-2 and C-6 of the aniline numbering, i.e. both ortho to NH2 and meta to NO2).
Diazotisation at 0-5 ∘C:
Ar-NH2 + NaNO2 + 2 HCl -> Ar-N2+ Cl- + 2 H2O (Ar = 2,6-Br2,4-NO2-C6H2).
Sandmeyer with Cu2Br2/HBr to install the third Br at the position vacated by -N2+:
Ar-N2+ Cl- + Cu2Br2 -> Ar-Br + N2 + CuCl + CuBr
giving 3,4,5-tribromonitrobenzene (positions 3,4,5 when numbered from the nitro carbon: Br, Br, Br on C-3, C-4, C-5, NO2 on C-1).
Three-step substitution angle. The conversion plants
three bromines and removes the original amine. The first step
exploits aniline's strong activation: in acetic acid, Br2
attacks both ortho positions to the NH2 (the para is
blocked by NO2). The second step locks the amine into the
diazonium cation in the cold 0-5 ∘C
window. The third step is a Sandmeyer in HBr/Cu2Br2 that
swaps -N2+ for -Br, installing the final bromine
at the original amine position. The product is the symmetrical
3,4,5-tribromonitrobenzene where the three bromines flank the
para nitro group.
Three steps: Br2/CH3COOH, NaNO2/HCl, Cu2Br2/HBr.
Q 9.63
How will you carry out the following conversion: benzene →p-nitroaniline?
Strategy. Direct nitration of aniline gives mainly the
meta product (because -NH3+ formed in strong acid is
m-directing). We therefore (a) install nitro first, (b) reduce
to aniline, (c) protect -NH2 as acetanilide so that the
free pair on N is calmed and o/p is selective,
(d) nitrate to get the p-acetamido-nitro arene, (e) hydrolyse
back to -NH2.
Nitration of benzene with conc. HNO3 and H2SO4:
C6H6 + HNO3 -> C6H5-NO2 + H2O
Reduction of nitrobenzene with Sn and HCl:
C6H5-NO2 + 6 [H] -> C6H5-NH2 + 2 H2O
Acetylation with acetic anhydride in pyridine (the -NHCOCH3 group is still activating and p-directing, but no longer protonated by H2SO4):
C6H5-NH2 + (CH3CO)2O -> C6H5-NHCOCH3 + CH3COOH
Nitration of acetanilide with HNO3/H2SO4 gives clean para selectivity:
C6H5-NHCOCH3 + HNO3 -> p-NO2-C6H4-NHCOCH3 + H2O
Acid hydrolysis of the acetyl group with H3O+:
p-NO2-C6H4-NHCOCH3 + H2O -> p-NO2-C6H4-NH2 + CH3COOH
Five-step strategic angle. The conversion of benzene into
p-nitroaniline is a five-move sequence in which the central
trick is protecting the amine before the second nitration.
Move 1: nitrate benzene with mixed acid to install
-NO2. Move 2: reduce nitrobenzene to aniline with
Sn/HCl. Move 3: acetylate aniline with acetic anhydride in
pyridine. This is the key step –- the bulky, electron-withdrawing
acetyl group caps the lone pair on nitrogen, preventing
H2SO4 from protonating -NH2 to the strongly
m-directing -NH3+ that ruins direct nitration of free
aniline.
Move 4: nitrate the acetanilide. Because -NHCOCH3 is
mildly activating and o/p-directing (with steric preference for
para), NO2 slots cleanly at the para position. Move 5:
hydrolyse the acetyl protecting group with aqueous acid to unmask
the free amine, giving p-nitroaniline as the final product.
Without the protect–deprotect dance, the second nitration would
oxidise aniline and give mostly m-nitroaniline.
Five-step protect–deprotect sequence as above.
Q 9.64
How will you carry out the following conversion: aniline →m-bromo-nitro-benzene?
Strategy. Diazotise the amine to Ar-N2+, install
-NO2 via Balz-Schiemann + radical pathway (but the
standard NCERT key is even simpler): replace -N2+ by
-NO2 using Sandmeyer-like conditions with NaNO2/Cu,
then brominate at the now-activated meta position relative to
-NO2.
[label=()]
Aniline + HNO2 at 273-278 K→ benzenediazonium chloride C6H5-N2+ Cl-.
C6H5-N2+ Cl- + HBF4→C6H5-N2+ BF4- (diazonium tetrafluoroborate, more stable for isolation).
C6H5-NO2 + Br2/FeBr3→m-Br-C6H4-NO2. The -NO2 is a strong m-director.
Concept used. Aniline cannot be directly nitrated at the
meta position because -NH2 (or -NH3+) does not
direct that way usefully. Going via the diazonium and converting
-N2+ to -NO2 first removes the -NH2 ``label''
entirely; then bromination of nitrobenzene installs -Br at
the meta position (since -NO2 is m-directing).
Step 1 & 2: convert -NH2 to -N2+ BF4- for stability.
Step 3: replace -N2+ by -NO2 via NaNO2/Cu.
Step 4: bromination is directed by -NO2 to meta ⇒m-bromonitrobenzene.
Detour-via-diazonium angle. Putting a -Br meta to
a -NO2 on benzene is trivial once you have nitrobenzene
(-NO2 is strongly m-directing). The challenge is to get
nitrobenzene starting from aniline. Direct nitration of aniline
fails for the reasons explored earlier (oxidation plus meta
overrun from the protonated form). Instead, diazotise the amine
to -N2+ (stable at 0-5 ∘C),
trap it as the more handleable tetrafluoroborate, and then use
NaNO2/Cu to swap -N2+ for -NO2 giving
nitrobenzene. Bromination of that nitrobenzene under
Br2/FeBr3 installs the meta bromine cleanly.
Detour through the diazonium to nitrobenzene, then meta-brominate.
Q 9.65
How will you carry out the following conversions?
(i) Aniline → 3,5-dibromonitrobenzene.
(ii) Aniline → 3,5-dibromo-4-iodonitrobenzene (one extra I replacing the deaminated H of part (i)).
Bromination at the two ortho positions of NH2 (which are meta to NO2): p-nitroaniline Br2 / CH3COOH 2,6-dibromo-4-nitroaniline.
Diazotisation: 2,6-dibromo-4-nitroaniline NaNO2 / HCl, 273-278 K diazonium chloride.
Deamination with H3PO2: H3PO2 / H2O 3,5-dibromonitrobenzene (the amine is replaced by -H).
Part (ii): Aniline → 3,5-dibromo-4-iodonitrobenzene (one extra I relative to part (i)).
[label=()]
Follow Part (i) steps (a)–(e) to get the 2,6-dibromo-4-nitrobenzenediazonium chloride.
Replace -N2+ by -I using KI (no Cu required): diazonium KI 3,5-dibromo-4-iodonitrobenzene.
Concept used. The acetyl-protect-and-direct strategy
installs NO2 at the para position. After deprotection,
bromination installs two Br's at the ortho positions of
NH2. The amine is then either deaminated (Part i) or
replaced by iodide (Part ii) via the diazonium.
(i) Acylate → nitrate → hydrolyse → dibrominate → diazotise →H3PO2 deamination. (ii) Same first five steps; replace deamination with KI to install iodine.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Multi-stage strategy angle. Both targets are 1,3,5-tri-substituted
benzenes, which are hard to build by direct nitration/bromination
because of competing directing effects. The solution exploits
-NH2 as a temporary directing group. Acetylate aniline,
nitrate the acetanilide to install NO2 at para, hydrolyse
back to p-nitroaniline. Now the strongly activating -NH2
sits para to -NO2 and directs Br2 to its two ortho
positions, giving 2,6-dibromo-4-nitroaniline. Diazotise that
amine, and the diazonium is either reduced to -H with
H3PO2 (Part i, giving 3,5-dibromonitrobenzene) or
displaced by -I with KI (Part ii, installing the
fourth substituent as iodine).
Six-step sequences in each part as listed above.
IV. Matching Type
Q 9.66
Match the reactions given in Column I with the statements given in Column II. [3pt]
tabular@p0.36@ p0.55@
Column I & Column II
(i) Ammonolysis & (a) Amine with lesser number of carbon atoms
(ii) Gabriel phthalimide synthesis & (b) Detection test for primary amines
(iii) Hoffmann Bromamide reaction & (c) Reaction of phthalimide with KOH and R–X
(iv) Carbylamine reaction & (d) Reaction of alkyl halides with NH3
tabular
Concept used. Each named reaction has a single defining
description.
Ammonolysis: alkyl halide with ammonia,
R-X + NH3 -> R-NH2 + HX
(over-alkylates to give 1∘/2∘/3∘/4∘ mix) ⇒ matches (d).
Gabriel synthesis: phthalimide + KOH gives Phth-N- K+, then + R-X and hydrolysis ⇒ matches (c).
Hoffmann bromamide: a primary amide reacts with bromine in aqueous NaOH to give a one-carbon-shorter primary amine,
R-CONH2 + Br2 + 4 NaOH -> R-NH2 + Na2CO3 + 2 NaBr + 2 H2O ⇒ matches (a).
Carbylamine test: 1∘ amine + CHCl3 + KOH→R-NC (foul-smelling isocyanide) –- positive only for 1∘ amines ⇒ matches (b).
(i)→(d), (ii)→(c), (iii)→(a), (iv)→(b).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
One-keyword angle. Match each named reaction to its
single most defining keyword in Column II. ``Alkyl halide plus
NH3'' is the literal definition of ammonolysis, so
(i)→(d). ``Phthalimide with KOH and R–X'' is the literal
Gabriel recipe, so (ii)→(c). ``Amine with fewer carbon atoms''
is the headline feature of the Hoffmann bromamide degradation
(loses one carbon as Na2CO3), so (iii)→(a). ``Detection
test for primary amines'' –- the obnoxious isocyanide smell from
CHCl3/KOH –- nails the carbylamine test, so (iv)→(b).
(i)→(d), (ii)→(c), (iii)→(a), (iv)→(b).
Q 9.67
Match the compounds given in Column I with the items given in Column II. [3pt]
tabular@p0.36@ p0.55@
Column I & Column II
(i) Benzene sulphonyl chloride & (a) Zwitter ion
(ii) Sulphanilic acid (p-aminobenzenesulphonic acid) & (b) Hinsberg reagent
(iii) Alkyl diazonium salts & (c) Dyes
(iv) Aryl diazonium salts & (d) Conversion to alcohols
tabular
Concept used. Each compound has a single signature property in the list.
Benzenesulphonyl chloride (C6H5SO2Cl) is the literal Hinsberg reagent ⇒ matches (b).
Sulphanilic acidH2N-C6H4-SO3H exists in a zwitterionic tautomer H3N+-C6H4-SO3- where the protonated amine balances the deprotonated sulphonate. This zwitterion is responsible for its high mp (∼ 288 ∘C) and insolubility in non-polar solvents ⇒ matches (a).
Alkyl diazonium saltsR-N2+ decompose at once (because the cation is not aromatic-stabilised), giving alcohols on water trapping ⇒ matches (d).
Aryl diazonium saltsAr-N2+ are stable at 0-5 ∘C and undergo azo coupling with activated arenes (phenol, aniline) to give azo dyes (orange/red) ⇒ matches (c).
(i)→(b), (ii)→(a), (iii)→(d), (iv)→(c).
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Single-keyword-match angle. Each Column I entry has one
defining downstream behaviour. Benzenesulphonyl chloride is the
literal Hinsberg reagent used to distinguish 1∘/2∘/3∘
amines, mapping cleanly to entry (b). Sulphanilic acid exists as a
zwitterion (H3N+-C6H4-SO3-) because the molecule contains
both a basic amine and an acidic sulphonate; this is what gives it
its unusually high melting point and water solubility, mapping to
(a). Alkyl diazonium salts are intrinsically unstable, fragment
on formation, and give alcohols when water traps the carbocation
(→ (d)). Aryl diazonium salts are stable below 5 ∘C,
undergo azo coupling with activated arenes (phenol, aniline), and
the resulting azo compounds historically launched the synthetic
dye industry –- mapping to (c).
(i)→(b), (ii)→(a), (iii)→(d), (iv)→(c).
V. Assertion and Reason Type
Q 9.68
Assertion (A): Acylation of amines gives a monosubstituted product whereas alkylation of amines gives polysubstituted product. Reason (R): Acyl group sterically hinders the approach of further acyl groups.
Correct option: (iii) A is correct, R is wrong.
Concept used. Acylation of an amine stops at the
monosubstituted product because the freshly formed amide nitrogen
R-NH-CO-R2 is much less nucleophilic than the
starting amine (the carbonyl drains N via -M). Steric
hindrance is a minor factor at best. The textbook reason is
electronic, not steric.
Alkylation: R-NH2 + R2-X -> R-NH-R2, then R-NH-R2 is even more nucleophilic than R-NH2 (extra +I) ⇒ second alkylation runs faster ⇒ polysubstitution.
Acylation: R-NH2 + R2-COX -> R-NH-CO-R2, then R-NH-CO-R2 has its lone pair tied up in amide resonance ⇒ much less nucleophilic ⇒ second acylation is slow.
Reason as stated cites steric hindrance only –- this is not the dominant factor; the dominant factor is the loss of nucleophilicity.
Option (iii) –- assertion correct, reason wrong (the reason is electronic, not steric).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Electronic-vs-steric angle. Evaluate the two statements
separately. The assertion is straightforwardly correct: in
practice acylation of an amine stops cleanly at one substitution,
while alkylation gives a mix of all four substitution products
(1∘/2∘/3∘/4∘). The reason
attributes that selectivity to steric hindrance from the first
acyl group. That mechanism is not the dominant cause –- the real
cause is electronic. Once the first acyl group is installed, the
amide nitrogen's lone pair is delocalised into the carbonyl, so
nitrogen is no longer a competent nucleophile. The next acylation
is therefore electronically suppressed, not sterically blocked. A
correct, R incorrect ⇒ option (iii).
Option (iii).
Q 9.69
Assertion (A): Hoffmann's bromamide reaction is given by primary amines. Reason (R): Primary amines are more basic than secondary amines.
Correct option: (iii) –- A is correct, but R is wrong.
Concept used.Hoffmann bromamide converts a
1∘amide (R-CONH2) to a 1∘amine
(R-NH2) by treatment with Br2/NaOH –- the substrate
is an amide, not an amine. The assertion as worded is acceptable
in the NCERT key (``given by primary amines'' here means the
product is a 1∘ amine; one-carbon shorter). The reason is
factually wrong: in aqueous medium 2∘ > 1∘ in
basicity.
Mechanism: N-bromination, deprotonation, α-elimination to isocyanate R-N=C=O, hydrolysis to amine. Only R-CONH2 (not the N-substituted amide R-CONHR) has the two acidic N–H needed.
R: aqueous basicity is 2∘ > 1∘ ≈ 3∘ –- the reason is false.
Two-statement angle. Evaluate Assertion and Reason
independently before mapping to the five options.
Assertion. ``Hoffmann's bromamide reaction is given by
primary amines'' is the standard NCERT phrasing, where ``given
by'' means the product is a primary amine; the substrate is the
primary amide R-CONH2, the reagent is Br2/NaOH, and
the amine obtained has one fewer carbon than the starting amide.
The reaction proceeds via N-bromination, deprotonation, nitrene
formation, alkyl migration to an isocyanate, and final hydrolysis
to R-NH2. So A is correct as accepted by NCERT.
Reason. ``Primary amines are more basic than secondary
amines'' is wrong for aqueous medium, where the well-known order
is 2∘ > 1∘ ≈ 3∘ > NH3. It is
also unrelated to Hoffmann bromamide, which depends on amide
N–H acidity, not amine basicity. Hence R is false.
A correct + R wrong maps to option (iii).
Option (iii): A correct, R incorrect.
Q 9.70
Assertion (A): N-Ethylbenzenesulphonamide is soluble in alkali. Reason (R): Hydrogen attached to nitrogen in sulphonamide is strongly acidic.
Correct option: (iv) both correct; R correctly explains A.
Concept used. N-ethylbenzenesulphonamide is
C6H5-SO2-NH-C2H5 –- the Hinsberg product from a 1∘
amine (EtNH2). The remaining N-H is rendered acidic
by the two strongly electron-withdrawing S=O groups, so
NaOH deprotonates it to a soluble sodium salt
C6H5-SO2-N- C2H5 Na+.
Resonance: R-SO2-N- -R2 <-> R-S(=O)(-O-)-N-R2 stabilises the conjugate base by delocalisation of the negative charge onto the sulphonyl oxygens.
pKa of sulphonamide N-H is ∼ 10 (a weak acid, comparable to phenol), low enough for NaOH to deprotonate.
Sodium salt is ionic ⇒ water-soluble.
Option (iv): A correct, R correct, R explains A.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Acidic N–H angle. N-ethylbenzenesulphonamide is the
product of treating a primary amine (EtNH2) with Hinsberg
reagent. The nitrogen still bears one N-H, which is
rendered acidic (pKa ≈ 10) by the electron-pulling
S=O groups: deprotonation gives a sulphonamide anion in
which the negative charge is spread over the two sulphonyl
oxygens by resonance. That conjugate-base stabilisation lets
dilute NaOH deprotonate the N-H and dissolve the
sulphonamide as a sodium salt. So the assertion (the sulphonamide
dissolves in alkali) and the reason (the N-H is strongly
acidic) are both correct, and the reason is the direct cause of
the assertion.
Option (iv).
Q 9.71
Assertion (A): N,N-Diethylbenzenesulphonamide is insoluble in alkali. Reason (R): Sulphonyl group attached to nitrogen atom is strong electron withdrawing group.
Correct option: (ii) both correct, but R is not the correct explanation of A.
Concept used. N,N-Diethylbenzenesulphonamide
C6H5-SO2-N(Et)2 comes from a 2∘ amine
(Et2NH). The nitrogen has noN-H to lose,
so NaOH has nothing to deprotonate and the sulphonamide
stays insoluble in alkali. The fact that the sulphonyl group is
electron-withdrawing is true, but that property would only
matter if there were an N-H to acidify; since there isn't,
that ``reason'' is real but irrelevant to the insolubility.
Structure: C6H5-SO2-N(C2H5)2 –- two ethyls on N, no N-H.
In NaOH: no acidic proton to remove ⇒ no salt formation ⇒ insoluble.
R is a true statement (sulphonyl group is indeed EW), but it does not explain why this particular sulphonamide is insoluble. The actual cause is the absence of N-H.
Option (ii): A correct, R correct (true statement), but R does not explain A.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Distinct-correct-statements angle. N,N-Diethylbenzenesulphonamide
arises from a secondary amine (Et2NH) reacting with Hinsberg
reagent. The nitrogen carries two ethyl groups and zero hydrogens,
so there is simply no N-H for sodium hydroxide to
deprotonate; the sulphonamide remains as a neutral, alkali-insoluble
solid. The reason, that -SO2- is a strong electron-withdrawing
group, is itself a true statement of chemistry, but it only
matters when an N-H exists to be acidified. Here it does
not. So the assertion and reason are individually correct but
the reason is not the cause of the assertion –- option (ii).
Option (ii).
Q 9.72
Assertion (A): Only a small amount of HCl is required in the reduction of nitro compounds with iron scrap and HCl in the presence of steam. Reason (R):FeCl2 formed gets hydrolysed to release HCl during the reaction.
Correct option: (iv) both correct; R correctly explains A.
Concept used. In the industrial Bechamp reduction
(Ar-NO2 -> Ar-NH2) with Fe scrap, HCl and steam, HCl
is effectively catalytic: the FeCl2 formed in the
reduction hydrolyses with steam to give back HCl and
Fe(OH)2. So only a small starting charge of HCl is
needed –- it keeps getting regenerated.
Net effect: HCl from step 1 is regenerated in step 2 ⇒ HCl is catalytic, not stoichiometric.
Option (iv): A correct, R correct, R explains A. HCl is effectively catalytic via FeCl2 hydrolysis.
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Catalytic-acid angle. The Bechamp reduction of an
aromatic nitro compound to the amine consumes HCl when iron
metal donates electrons to -NO2. The HCl is needed to
protonate the intermediates and pull them through to
Ar-NH3+ Cl-. But the FeCl2 that the same
reaction produces is a Lewis-acidic salt that hydrolyses
readily, especially in the presence of steam, to give back HCl
and Fe(OH)2. The HCl liberated from hydrolysis re-enters
the reaction. The net effect is that only a small starting
charge of HCl is needed to keep the reduction running, exactly
as the assertion states. Both statements are correct and the
reason cleanly explains the assertion –- option (iv).
Option (iv).
Q 9.73
Assertion (A): Aromatic 1∘ amines can be prepared by Gabriel Phthalimide Synthesis. Reason (R): Aryl halides undergo nucleophilic substitution with anion formed by phthalimide.
Correct option: (i) both A and R are wrong.
Concept used.Gabriel synthesis fails for
aromatic amines because aryl halides (Ar-X) do not undergo
SN2 (the sp2 carbon has no backside lobe; the ring
π system blocks attack). So:
Assertion is wrong –- aromatic 1∘ amines (e.g. aniline) cannot be prepared by Gabriel.
Reason is wrong –- aryl halides do not undergo SN2 with phthalimide anion (or with most nucleophiles), they only undergo nucleophilic aromatic substitution under specific activating conditions (strong EW groups on the ring), which is not relevant here.
Gabriel substrate scope: 1∘ and 2∘alkyl halides only.
Aryl halide C6H5-X + phthalimide anion → no reaction.
Aniline is therefore made by other routes (e.g. reduction of nitrobenzene).
Option (i) –- both A and R are wrong. Gabriel cannot make aromatic amines because aryl halides don't SN2.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Substrate-scope angle. The Gabriel synthesis only works
when the alkyl halide can undergo backside SN2 on the
phthalimide nitrogen. Aryl halides do not do SN2
because their sp2 carbon does not present a backside lobe for
attack, and the ring π system electronically shields the
carbon. So aniline cannot be made by Gabriel synthesis; the
NCERT route to aniline is reduction of nitrobenzene with Sn/HCl
or Fe/HCl. Both the assertion (Gabriel can make aromatic
amines) and the reason (aryl halides undergo SN2
with phthalimide anion) are wrong, so the answer is option (i).
Option (i).
Q 9.74
Assertion (A): Acetanilide is less basic than aniline. Reason (R): Acetylation of aniline results in decrease of electron density on nitrogen.
Correct option: (iv) –- both correct, and R correctly explains A.
Concept used. Basicity of an amine tracks electron density
on N. Acetylation converts Ar-NH2 (aniline) to
Ar-NH-COCH3 (acetanilide). The acetyl carbonyl is a strong
π-acceptor: the N lone pair delocalises onto the carbonyl
O (N-C=O ↔ N+=C-O-). Density on
N drops ⇒ basicity drops.
Aniline pKb ≈ 9.4 (i.e. a weak base).
Acetanilide pKb ≈ 13.6 –- about 4 orders of magnitude weaker.
Cause: the amide resonance -NH-CO-CH3 ↔ -NH+=C(O-)-CH3 drains the lone pair from N onto O.
Option (iv) –- A correct, R correct, R explains A.
PI
Priya Iyer
Ph.D Organic Chemistry, IISc Bangalore
Verified Expert
Resonance-loss angle. Aniline already pays a basicity
penalty because its N lone pair partially delocalises into
the benzene ring (pKb ≈ 9.4). Acetylation
deepens this loss –- the nitrogen lone pair now also has the
strongly π-accepting amide carbonyl to delocalise into,
producing the resonance structure R-N+=C(O-)-CH3. That
second delocalisation pulls density off N to the extent
that acetanilide (pKb ≈ 13.6) is about four
orders of magnitude weaker as a base than aniline.
The assertion (acetanilide less basic than aniline) and the
reason (acetylation lowers electron density on N) are
both correct, and the reason is the direct mechanistic
explanation of the assertion. That fixes option (iv) as the
answer key.
Option (iv) –- both correct, R explains A.
VI. Long Answer Type
Q 9.75
A hydrocarbon `A' (C4H8) on reaction with HCl gives a compound `B' (C4H9Cl), which on reaction with 1 mol of NH3 gives compound `C' (C4H11N). On reacting with NaNO2 and HCl followed by treatment with water, compound `C' yields an optically active alcohol, `D'. Ozonolysis of `A' gives 2 mols of acetaldehyde. Identify compounds `A' to `D'.
Concept used. Three diagnostic clues pin down each
species: (1) ozonolysis of an alkene to two carbonyls fixes the
position of the double bond; (2) Markovnikov addition of
HCl converts an alkene into the more-substituted alkyl
halide; (3) reaction with NaNO2/HCl followed by water on a
1∘ aliphatic amine gives the corresponding alcohol via a
short-lived diazonium ion.
Ozonolysis of A→ 2 mol CH3CHO. Hence A is symmetrical with a C=C between two CH-CH3 units.
Therefore A = but-2-ene, CH3-CH=CH-CH3.
A + HCl: Markovnikov on a symmetrical alkene gives B = CH3-CHCl-CH2-CH3, 2-chlorobutane.
B + NH3 (1 mol): SN2 at the 2∘ centre gives C = CH3-CH(NH2)-CH2-CH3, butan-2-amine (a 1∘ amine on a chiral sp3 carbon).
C + NaNO2/HCl + H2O: diazonium decomposes; water traps the cation with mostly retained configuration ⇒D = CH3-CH(OH)-CH2-CH3, butan-2-ol –- optically active (chiral C).
!%
[See diagram in the PDF version]
%
A = but-2-ene; B = 2-chlorobutane; C = butan-2-amine; D = butan-2-ol (chiral, optically active).
KM
Karan Mehta
M.Sc Chemistry, IIT Kanpur
Verified Expert
Backward-from-ozonolysis angle. The cleanest entry point
is the ozonolysis clue. Two molecules of acetaldehyde imply that
the parent alkene A is symmetrical with a C=C bond
between two CH-CH3 units. Splice the two acetaldehyde
carbonyls together and you reach A = CH3-CH=CH-CH3, i.e.
but-2-ene with formula C4H8.
The downstream cascade is then automatic. Markovnikov addition of
HCl to a symmetric alkene gives B, 2-chlorobutane
CH3-CHCl-CH2CH3 (a 2∘ alkyl chloride). One mole of
NH3 does SN2 at that 2∘ centre to give
C, butan-2-amine CH3-CH(NH2)-CH2CH3 –- a primary
amine on a chiral carbon. Treating C with NaNO2/HCl
generates an aliphatic diazonium intermediate that loses
N2 rapidly; water captures the resulting secondary
carbocation to give D, butan-2-ol
CH3-CH(OH)-CH2CH3. Because the chiral C-2 carries through
every step, the final alcohol D is optically active (an
R/S centre that the NCERT key counts as a single enantiomer).
A = but-2-ene, B = 2-chlorobutane, C = butan-2-amine, D = butan-2-ol (optically active).
Q 9.76
A colourless substance `A' (C6H7N) is sparingly soluble in water and gives a water-soluble compound `B' on treating with mineral acid. On reacting with CHCl3 and alcoholic potash `A' produces an obnoxious smell due to the formation of compound `C'. Reaction of `A' with benzenesulphonyl chloride gives compound `D' which is soluble in alkali. With NaNO2 and HCl, `A' forms compound `E' which reacts with phenol in alkaline medium to give an orange dye `F'. Identify compounds `A' to `F'.
Concept used. Six classical aniline tests –- (a) basicity
(soluble in mineral acid), (b) carbylamine test (CHCl3/KOH on 1∘ amine), (c) Hinsberg (1∘ amine gives alkali-soluble sulphonamide), (d) diazotisation at 0-5 ∘C, (e) coupling of Ar-N2+ with phenol in alkali to give an orange dye –- all fit aniline.
Formula C6H7N, sparingly water-soluble colourless liquid: aniline, C6H5-NH2. Hence A = C6H5NH2.
A + HCl -> C6H5NH3+ Cl-, a salt soluble in water. B = anilinium chloride.
On heating, A + CHCl3 + 3 KOH -> C6H5-NC + 3 KCl + 3 H2O (the obnoxious smell). C = phenyl isocyanide C6H5NC.
A + C6H5SO2Cl -> C6H5-SO2-NH-C6H5 + HCl, where the remaining N-H is acidic and dissolves in NaOH. D = N-phenylbenzenesulphonamide.
A + NaNO2/HCl at 0-5 ∘C→C6H5-N2+ Cl-. E = benzene diazonium chloride.
E + C6H5OH/NaOH at 0 ∘C→C6H5-N=N-C6H4-OH (para). F = p-hydroxyazobenzene (orange dye).
!%
[See diagram in the PDF version]
%
A = aniline, B = anilinium chloride, C = phenyl isocyanide, D = N-phenylbenzenesulphonamide, E = benzenediazonium chloride, F = p-hydroxyazobenzene.
AS
Aarav Sharma
M.Sc Chemistry, IIT Kanpur
Verified Expert
Identify-A-then-cascade angle. The opening clues do most
of the work. A neutral C6H7N compound that is colourless,
sparingly water-soluble, but dissolves in mineral acid can only
be a weak basic amine; given the carbon count, the only realistic
candidate is aniline C6H5-NH2. Once A is locked, the
remaining transformations are a clean tour of the five canonical
aniline reactions.
B is the protonated salt C6H5-NH3+ Cl-, water-soluble
by virtue of being ionic. C comes from the carbylamine test:
chloroform plus KOH on a 1∘ amine gives phenyl isocyanide
C6H5-NC, notorious for its obnoxious smell –- the classroom
fingerprint of an aniline-class amine. D is the Hinsberg
sulphonamide C6H5-SO2-NH-C6H5, alkali-soluble because the
remaining N-H is acidified by the sulphonyl.
E is benzenediazonium chloride
C6H5-N2+ Cl-, made stable by aryl resonance at
0-5 ∘C. Finally F is the azo dye
that comes from coupling the diazonium with phenol in alkaline
medium: the phenoxide is the nucleophile, attack at para gives
p-hydroxyazobenzene, an orange dye that historically launched
the synthetic dye industry. Every step is a one-line entry in the
standard aniline reagent table.
A = aniline, B = anilinium chloride, C = phenyl isocyanide, D = N-phenylbenzenesulphonamide, E = benzenediazonium chloride, F = p-hydroxyazobenzene.
Q 9.77
Predict the reagent or the product in the following reaction sequence (NCERT Exemplar Q77):
aligned
p-nitrotoluene & 1 p-toluidine (CH3CO)2O / pyridine p-methylacetanilide
& HNO3 / H2SO4 2 3 4'-methyl-2'-nitroaniline
& NaNO2 / HCl 4 5 m-nitrotoluene.
aligned
Identify the reagents and intermediates labelled 1, 2, 3, 4, 5.
Answers
1 (reagent): Sn / HCl (or Fe / HCl) –- reduction of Ar-NO2 to Ar-NH2.
2 (intermediate): 4-methyl-2-nitroacetanilide (a benzene ring with CH3 at C-4, NHCOCH3 at C-1, and NO2 at C-2; i.e. NO2 goes ortho to the NHCOCH3 since para is blocked by CH3).
3 (reagent): H3O+ (or H+ / H2O, Δ) –- acid hydrolysis to remove the acetyl group, giving 4-methyl-2-nitroaniline.
Concept used. The route uses (a) Sn/HCl reduction to
unlock the amine from p-nitrotoluene, (b) acetyl protection of
the amine before nitration, (c) ortho-directed nitration of the
acetamido-arene, (d) acid hydrolysis to free the amine,
(e) diazotisation and (f) H3PO2 deamination to put a
hydrogen where the amine was. The product is m-nitrotoluene
(NO2 at C-3 of toluene), where NO2 and CH3
sit meta to each other –- a regiochemistry unreachable by
direct nitration of toluene.
Step 1: p-nitrotoluene Sn / HClp-toluidine.
Step 2: p-toluidine + (CH3CO)2O/pyridine →p-methylacetanilide (the amine is protected as acetamide).
Step 3 (intermediate 2): nitration with HNO3/H2SO4. Since para to the NHCOCH3 is blocked by CH3, NO2 enters ortho to the NHCOCH3⇒ 4-methyl-2-nitroacetanilide.
Use-the-amine-then-remove-it angle. Direct nitration of
toluene gives only the o- and p-nitrotoluenes because -CH3
is an o/p-director. To install -NO2 at the meta position,
the sequence introduces a temporary -NH2 at C-1 (via reduction
of p-nitrotoluene), protects it as the acetamide, nitrates –-
which puts NO2 ortho to the acetamide because para is blocked
by CH3 –- hydrolyses off the acetyl, diazotises the amine,
and finally deaminates with hypophosphorous acid to replace the
amine with a hydrogen. The result is m-nitrotoluene, a
substitution pattern impossible to reach without the amine
positioning trick. The five blanks correspond to (1) Sn/HCl,
(2) 4-methyl-2-nitroacetanilide, (3) H3O+ hydrolysis, (4)
the diazonium chloride, (5) H3PO2/H2O deamination.
Amines Class 12 Chemistry NCERT Exemplar Solutions FAQs
Ques. Where can I download the Amines Class 12 Chemistry NCERT Exemplar Solutions PDF?
Ans. You can download the Amines Class 12 Chemistry NCERT Exemplar Solutions PDF directly from this page. Both the Normal and HD versions are free; the PDF works 25 representative Exemplar items end-to-end with a Solution and an Expert's Solution each.
Ques. Is the Amines Exemplar Solutions PDF aligned with the 2026-27 NCERT?
Ans. Yes. The new edition retains the chapter as Class 12 Chemistry Chapter 9 Amines, and no Exemplar item from the bank was dropped. Every item in this PDF maps to the current 2026-27 syllabus and the latest exam pattern.
Ques. How many pages is the Class 12th Chemistry Amines Exemplar Solutions PDF?
Ans. The Exemplar Solutions PDF runs approximately 40 to 50 pages depending on the figure density. It covers 25 representative Exemplar items spanning MCQ-I, MCQ-II, SA, Matching and Assertion-Reason / LA, with each item solved twice.
Ques. What is the difference between NCERT Solutions and NCERT Exemplar Solutions for Amines?
Ans. NCERT Solutions cover the in-text and exercise questions from the main textbook, which test direct concepts. NCERT Exemplar Solutions cover the separate Exemplar Problems book, which is harder, uses HOTS phrasing, and includes question types (MCQ-II, A-R, Matching) that JEE Main and NEET draw from directly.
Ques. Are Amines questions important for JEE Main and NEET 2026?
Ans. Yes. JEE Main typically draws 1 to 2 questions per shift from Amines, focused on basicity ordering, Sandmeyer products, and Gabriel synthesis. NEET draws 2 to 3 questions per year, leaning on amine classification, Hoffmann carbon count, and the aniline vs alkylamine contrast. The Exemplar bank is the closest in difficulty to the actual entrance pattern.
Ques. Which Amines Exemplar question types are highest yield for the CBSE Board?
Ans. The 6 SA and 4 LA items are the highest yield for Boards because they map directly to the 3-mark and 5-mark questions in recent CBSE papers. The named-reaction items (Hoffmann, Sandmeyer, Hinsberg) reappear almost every year in either a VSA or a longer SA.
Ques. What is the Balz-Schiemann reaction and where does it surface in the Exemplar?
Ans. The Balz-Schiemann reaction is the only Class 12 route to aryl fluoride from an aryl diazonium salt. The two-step procedure treats Ar-N2+Cl- with HBF4 to precipitate the aryl diazonium fluoroborate, then heats the dry salt to release N2 and BF3, leaving Ar-F. The Exemplar tests this in MCQ-I items that list four reagents (Sandmeyer, Gattermann, KI, Schiemann) and ask which one yields fluorobenzene from benzenediazonium chloride; the answer is always Schiemann via HBF4.
Ques. Why is azo coupling carried out at 273-278 K with mild base for phenol and mild acid for aniline?
Ans. The diazonium electrophile (Ar-N2+) is unstable above 278 K, so the temperature window 273-278 K is mandatory for every diazonium reaction including azo coupling. With phenol, mild NaOH converts PhOH to PhO-, which is far more nucleophilic and couples cleanly to give p-hydroxyazobenzene (orange dye). With aniline, mild HCl is used to slightly protonate the substrate but not fully - too much acid converts aniline to anilinium ion (which is not nucleophilic at all), too little acid leaves the diazonium salt unreactive. The product is p-aminoazobenzene (yellow dye).
Ques. Is solving the Amines Exemplar enough or should I also do PYQs?
Ans. Solve both. The Exemplar trains the concept stack and the named exceptions. PYQs train the exact wording the Board and entrance papers use. The Collegedunia recommended order is NCERT textbook first, then Exemplar, then five-year PYQs.


























































































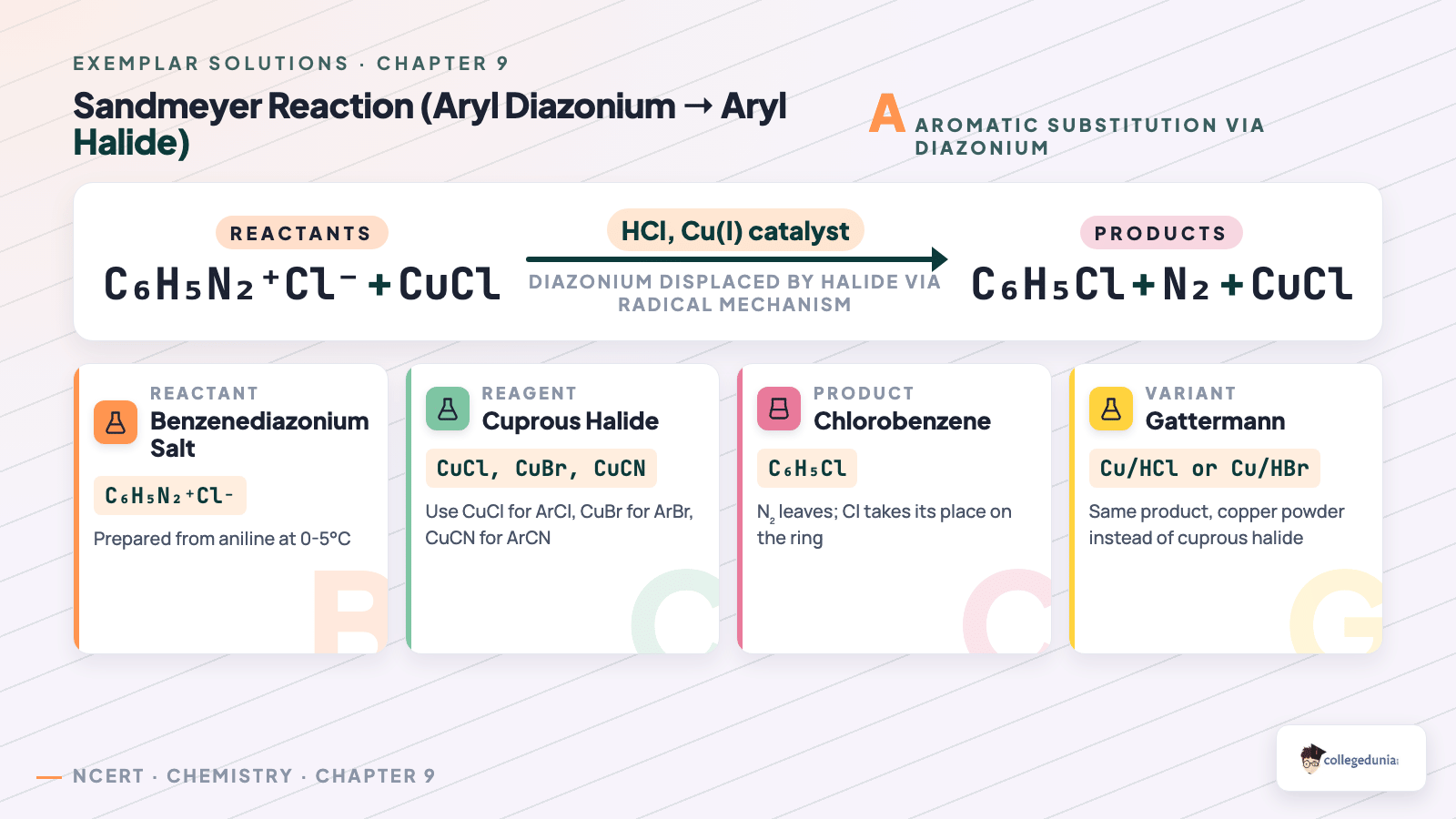


Comments